Le dermatosi autoimmuni bollose rappresentano un gruppo eterogeneo di malattie autoimmuni rare e talvolta gravi, che comprendono il pemfigo e le malattie pemfigoidi, l’epidermolisi bollosa acquisita e la dermatite erpetiforme di Duhring. Una caratteristica comune delle dermatosi autoimmuni bollose – ad eccezione della malattia di Duhring – sono gli autoanticorpi diretti contro le proteine strutturali della pelle e delle membrane mucose e responsabili della perdita dell’integrità cutanea [1].
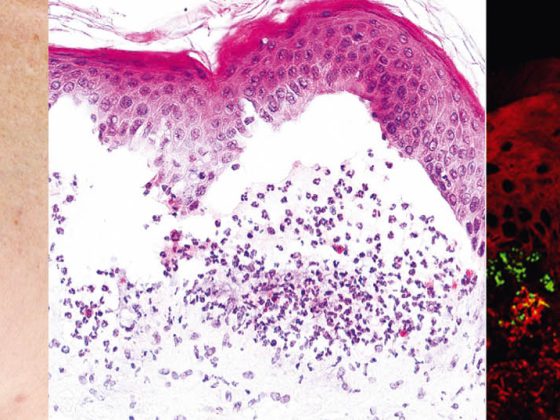
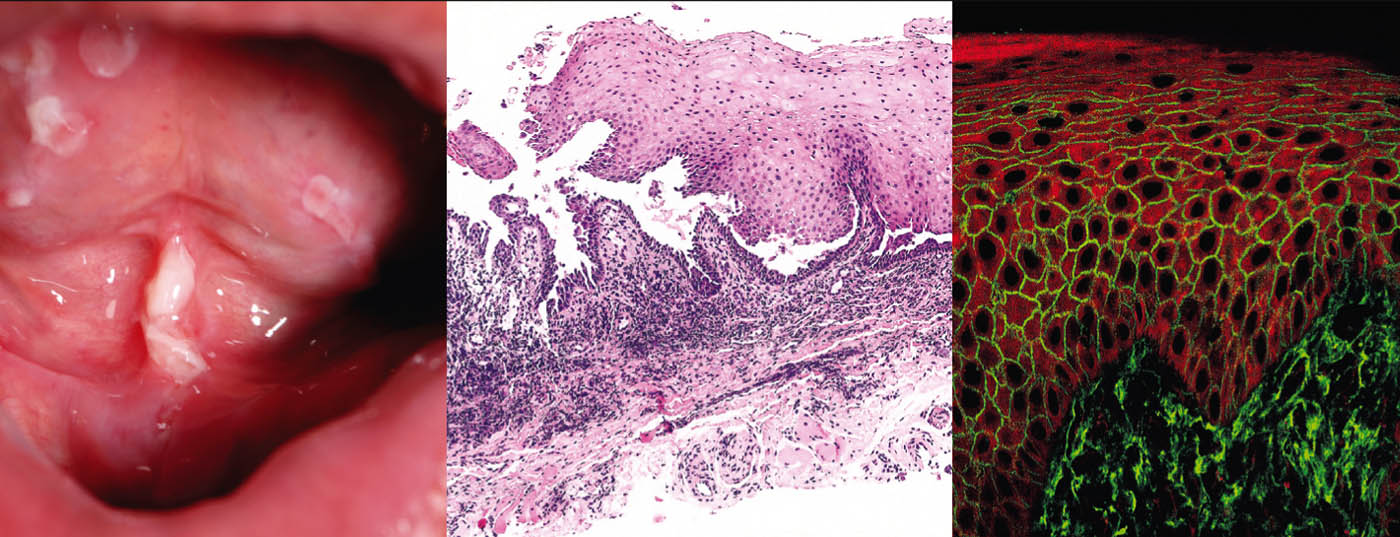
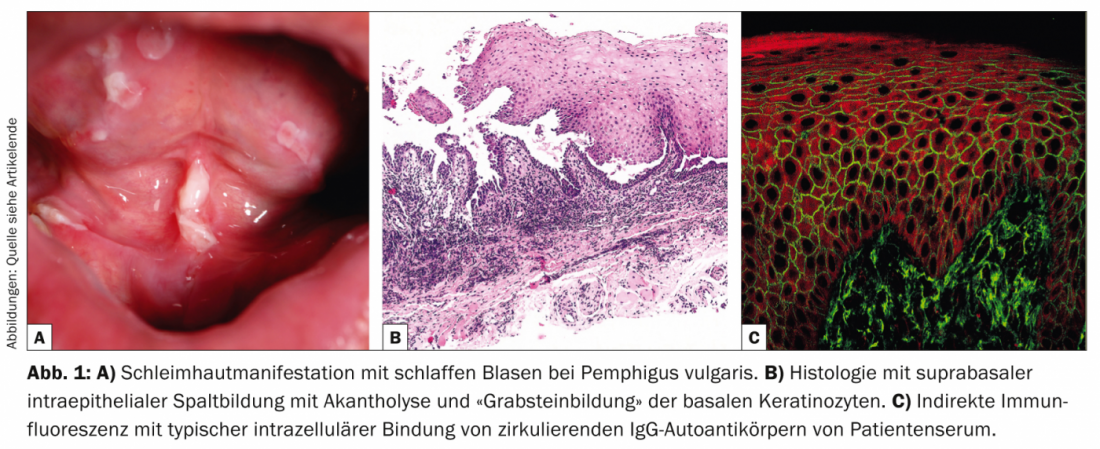
Il pemfigo vulgaris (Tab. 1, Fig. 1) si manifesta con vesciche flaccide generalizzate, mucocutanee, che si rompono molto rapidamente, per cui il quadro clinico è solitamente dominato da erosioni e croste.

Il coinvolgimento delle membrane mucose, soprattutto della bocca, che è molto doloroso, si verifica nella maggior parte dei pazienti e quindi la diagnosi viene fatta non di rado inizialmente dai dentisti. Dal punto di vista patogenetico, il pemfigo vulgaris è caratterizzato da autoanticorpi (AK) contro le proteine di adesione cellulare – le desmogleine 1 e 3. Nei pazienti con infestazione esclusivamente orale, le AK sono dirette contro la desmogleina 3. Dal punto di vista istopatologico, si osserva la formazione di una fessura intraepidermica soprabasale con acantolisi e “formazione di una pietra tombale” dei cheratinociti basali. L’immunofluorescenza diretta (DIF) mostra i tipici depositi intercellulari di IgG e C3 nell’epidermide. L’immunofluorescenza indiretta (IIF) dal siero del paziente conferma gli autoanticorpi IgG circolanti. Utilizzando l’ELISA, si può ottenere il rilevamento diretto della desmogleina 3 e della desmogleina 1 nel siero dei pazienti [2].
La terapia mira principalmente a ridurre la produzione di autoanticorpi. I corticosteroidi sistemici e altri immunosoppressori come l’azatioprina, il micofenolato mofetile, la ciclofosfamide o la ciclosporina rimangono le terapie standard. Altre opzioni sono la plasmaferesi o le immunoglobuline per via endovenosa (IVIG). Nei casi resistenti alla terapia, l’anticorpo anti-CD20 rituximab in particolare è un’alternativa promettente. Oltre al pemfigo volgare, il gruppo di malattie del pemfigo comprende anche il pemfigo foliaceo [3], il pemfigo erpetiforme [4], il pemfigo paraneoplastico [5] e il pemfigo IgA.
Pemfigoide bolloso
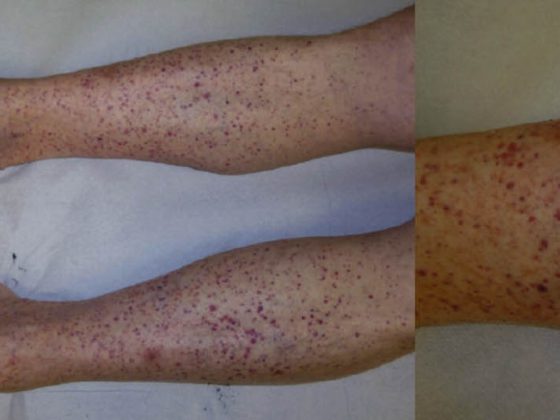
Con un’incidenza di 12,1 nuovi casi/milione/anno in Svizzera, il pemfigoide bolloso (Tab. 2, Fig. 2) è la malattia più frequente del gruppo pemfigoide e allo stesso tempo la dermatosi autoimmune formante vesciche più frequente di tutte [6].

Il pemfigoide bolloso si manifesta di solito in età avanzata ed è caratterizzato da vesciche rigonfie sulla pelle infiammata-arrossata o normale, che prudono intensamente. Spesso, questa malattia si presenta inizialmente senza vesciche e viene diagnosticata come eczema, orticaria o prurigo a causa del prurito pronunciato. Dal punto di vista patogenetico, la malattia è causata da autoanticorpi contro il BP 180 (noto anche come collagene di tipo XVII). L’istologia mostra vesciche subepidermiche con epidermide intatta e infiltrati prominenti ricchi di eosinofili. Nella DIF da pelle perilesionale, i depositi di IgG si trovano lungo la zona di giunzione dermo-epidermica (membrana basale), che può essere confermata nella IIF con il rilevamento degli anticorpi IgG che si legano alla membrana basale. Il livello sierico degli autoanticorpi contro il BP180 è correlato all’attività della malattia e può essere determinato durante il decorso per stabilire la necessità di un’ulteriore terapia.
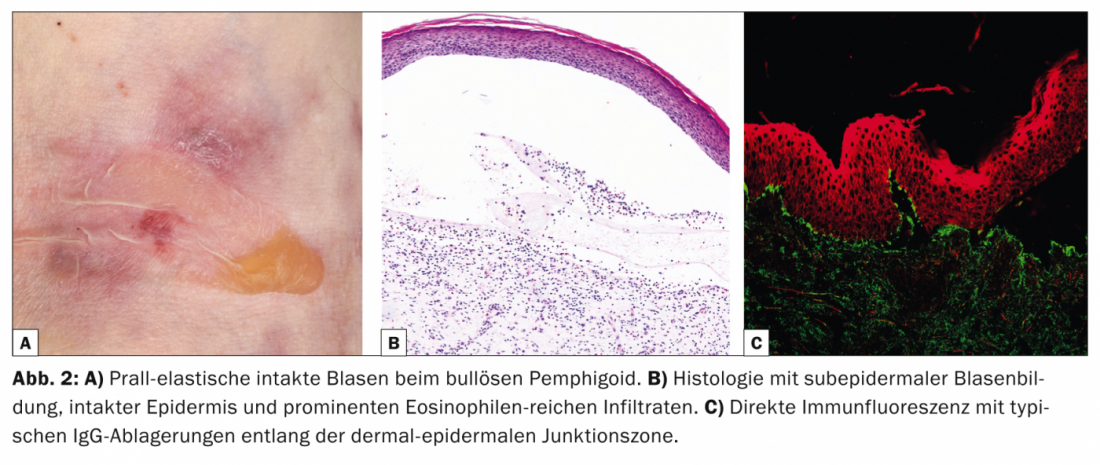
I corticosteroidi topici o sistemici rimangono la base della terapia. Altre opzioni includono le tetracicline con nicotinamidi, il dapsone o, nella malattia grave, gli immunosoppressori, in particolare l’azatioprina, nonché l’IVIG e il rituximab nei casi resistenti alla terapia.
IgA lineare, dermatosi bollosa
La dermatosi lineare IgA (LABD, Tab. 3, Fig. 3) è caratterizzata da vescicole e bolle pruritiche, generalizzate, accentuate sul lato estensore. La malattia si manifesta sia negli adulti che nei bambini.

Nell’infanzia, è la dermatosi bollosa autoimmune più comune e di solito è autolimitante [7]; negli adulti, la LABD è spesso associata a farmaci (vancomicina) [8]. Istologicamente, la LABD è simile alla malattia di Duhring in quanto è caratterizzata da vesciche subepidermiche, epidermide intatta e infiltrati ricchi di neutrofili nella giunzione dermo-epidermica. La DIF mostra depositi lineari di IgA nell’area della zona della membrana basale.
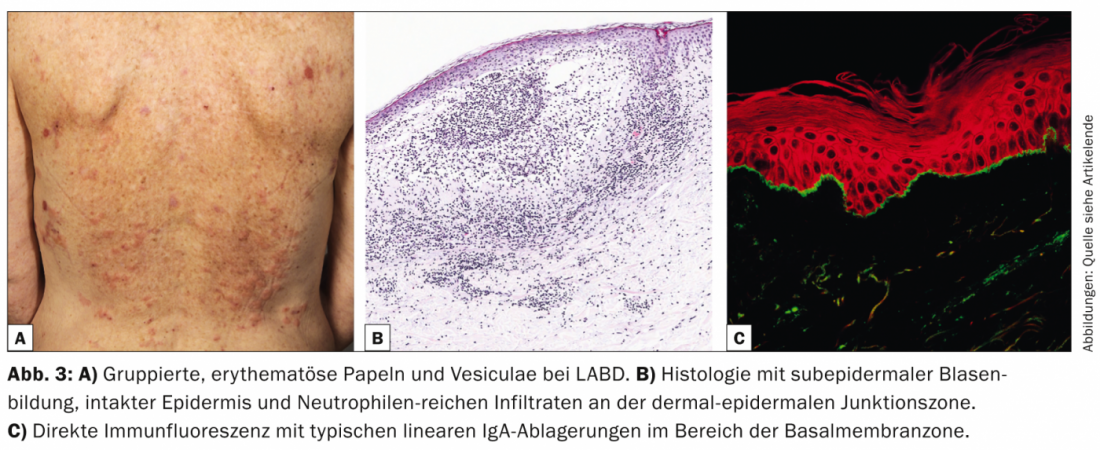
L’IIF può confermare la diagnosi rilevando gli autoanticorpi IgA circolanti che si legano al tetto della vescica. La maggior parte dei pazienti risponde alla terapia con dapsone o sulfapiridine. Sono stati descritti anche trattamenti di successo nei bambini e negli adulti con vari antibiotici (cicloxacillina, eritromicina, tetraciclina o trimetoprim-sulfametossazolo).
Epidermiolisi bollosa aquisita
L’epidermiolisi bollosa aquisita (EBA, Tab. 4, Fig. 4) è clinicamente suddivisa in una forma meccano-bollosa localizzata, spesso acromatica, non infiammatoria e cicatrizzata e in una variante generalizzata, infiammatoria e non cicatrizzata. La malattia si manifesta per lo più nella mezza età adulta ed è molto rara nei bambini.

Esiste una stretta associazione con la malattia infiammatoria intestinale [9]. Dal punto di vista patogenetico, l’EBA è caratterizzata dalla deposizione di autoanticorpi IgG contro il procollagene di tipo VII della fibrilla di ancoraggio. Istologicamente, il campione di routine mostra una vescica subepidermica con epidermide intatta. Nella DIF della pelle perilesionale, gli anticorpi IgG si trovano in bande lungo la zona di giunzione dermo-epidermica. Nell’IIF su cute divisa umana separata da NaCl, che è positiva nel 50% dei casi, gli autoanticorpi IgG o, meno frequentemente, IgA circolanti si legano al pavimento della vescica.
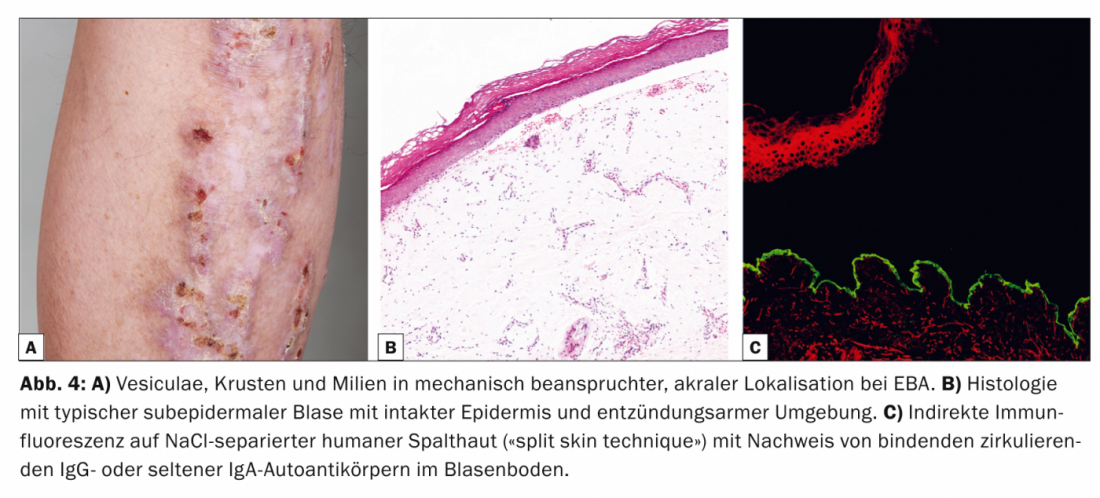
Il Western blot e l’ELISA possono essere usati in modo complementare per rilevare gli autoanticorpi IgG circolanti [10]. La terapia dei DMS è difficile, spesso insoddisfacente e mira principalmente a un effetto immunosoppressivo. Le attuali opzioni terapeutiche sono quindi principalmente i corticosteroidi sistemici, il dapsone o la colchicina. Inoltre, ci sono segnalazioni di una risposta positiva all’anticorpo anti-CD20 rituximab nei casi resistenti alla terapia.
Dermatite erpetiforme
La dermatite erpetiforme (DH, Tab. 5, Fig. 5) è una rara manifestazione cutanea dell’enteropatia sensibile al glutine (celiachia) e si presenta con papule eritematose e papulovescicole intensamente pruriginose, che si verificano principalmente sul lato estensore e sul sacro.

Dal punto di vista patogenetico, sia la DH che la celiachia sono associate al genotipo HLA-DQ2, in cui sono presenti autoanticorpi IgA contro la reticolina, l’endomisio e la transglutaminasi tissutale, rispettivamente. transglutaminasi epidermica. Istologicamente, c’è una formazione di fessure subepidermiche e infiltrati ricchi di neutrofili con formazione di microabscessi parziali [11].
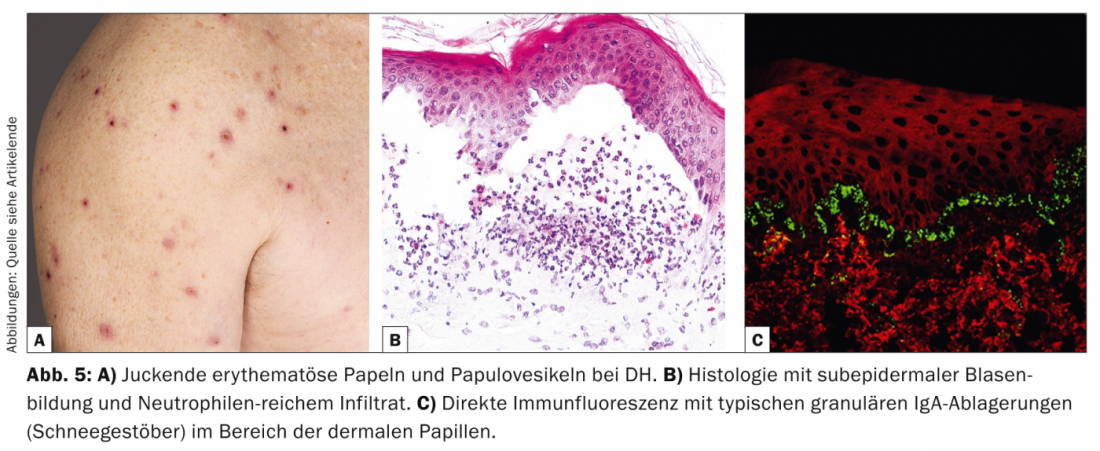
Istologicamente, la DH appare simile alla dermatosi bollosa lineare IgA, ma la DIF della pelle perilesionale mostra depositi granulari di IgA (fiocchi di neve) nelle papille dermiche. Dal punto di vista terapeutico, è in primo piano la dieta senza glutine, che migliora sia i disturbi gastroenterologici che i sintomi cutanei [12]. Le opzioni alternative sono il dapsone, le sulfapiridine o i corticosteroidi sistemici. Il dapsone migliora le lesioni cutanee ma non i sintomi intestinali.
Dott.ssa Emmanuella Guenova, MD
Fonti delle immagini:
Immagini cliniche: Archivio fotografico dell’Ospedale Universitario di Zurigo
Immagini istologiche: Dott.ssa Emmanuella Guenova, MD
Illustrazioni di immunofluorescenza: Birgit Fehrenbacher
Letteratura:
- Bolognia, JL, Jorizzo JL, Schaffer JV: Dermatologia. 2012: Elsevier Health Sciences UK.
- Chan LS: Malattie della pelle con vesciche. 2009: Taylor & Francis.
- Guenova E, et al: Tinea incognito nascosta sotto un pemfigo foliaceo apparentemente resistente al trattamento. Acta Derm Venereol 2008; 88(3): 276-277.
- Lebeau S, et al: Pemfigo erpetiforme: analisi del profilo autoanticorpale durante il decorso della malattia con cambiamenti nel fenotipo clinico. Clin Exp Dermatol 2010; 35(4): 366-372.
- Heizmann M, et al: Trattamento di successo del pemfigo paraneoplastico nel NHL follicolare con rituximab: rapporto di un caso e revisione del trattamento del pemfigo paraneoplastico nel NHL e nella LLC. Am J Hematol 2001; 66(2): 142-144.
- Marazza G, et al: Incidenza del pemfigoide bolloso e del pemfigo in Svizzera: uno studio prospettico di 2 anni. Br J Dermatol 2009; 161(4): 861-868.
- de las Heras MN: Dermatosi bollosa lineare IgA dell’infanzia: buona risposta al trattamento antibiotico. Clin Exp Dermatol 2014; 39(3): 395-397.
- Tashima S, et al: Un caso di dermatosi bollosa lineare IgA indotta da vancomicina con anticorpi IgA circolanti verso il dominio NC16a di BP180. Int J Dermatol 2014; 53(3): 207-209.
- Hundorfean G, Neurath MF, Sitaru C: Autoimmunità contro il collagene di tipo VII nella malattia infiammatoria intestinale. J Cell Mol Med 2010; 14(10): 2393-2403.
- Calabresi V, et al: Sensibilità di diversi test per la diagnosi sierologica dell’epidermolisi bollosa acquisita: analisi di una coorte di 24 pazienti italiani. J Eur Acad Dermatol Venereol 2014; 28(4): 483-490.
- Hall MA, Lanchbury JS, Ciclitira PJ: geni della regione HLA classe II e suscettibilità alla dermatite erpetiforme: le associazioni DPB1 e TAP2 sono secondarie a quelle della sottoregione DQ. Eur J Immunogenet 1996; 23(4): 285-296.
- Hervonen K, et al: Dermatite erpetiforme nei bambini: uno studio di follow-up a lungo termine. Br J Dermatol 2014 [Epub ahead of print].
CONCLUSIONE PER LA PRATICA
- In tutte le dermatosi autoimmuni bollose, oltre all’anamnesi, è importante l’esame dell’intero tegumento, compresa la pelle. L’ispezione delle membrane mucose e delle unghie è obbligatoria.
- Se si sospetta clinicamente o istologicamente una malattia autoimmune bollosa, la diagnosi deve essere confermata dal rilevamento degli autoanticorpi sottostanti (colorazione degli anticorpi legati al tessuto in immunofluorescenza diretta su sezione di tessuto o rilevamento nel siero mediante immunofluorescenza indiretta o ELISA).
- Nella terapia delle dermatosi autoimmuni bollose, si utilizzano ancora i classici immunosoppressori (ad esempio, corticosteroidi, azatioprina).
- Le opzioni terapeutiche più recenti sono gli anticorpi anti-CD20 (Rituximab), che portano a una riduzione degli autoanticorpi.
PRATICA DERMATOLOGICA 2014; 24(4): 6-10