La presenza di ILD nei pazienti con artrite reumatoide limita la prognosi, la sopravvivenza e la qualità di vita. La diagnosi precoce è di grande importanza, in modo da poter iniziare il trattamento in tempo e prevenire una progressione rilevante dal punto di vista prognostico. Le esacerbazioni acute dell’ILD possono complicare ulteriormente il decorso. Queste sono associate alla progressione della malattia o sono innescate secondariamente dalle infezioni.
La malattia polmonare interstiziale (ILD) è più comune nelle collagenosi, in particolare nella sclerosi sistemica (SSc), nelle miopatie autoimmuni, nella sindrome di Sjögren, nel lupus eritematoso sistemico (LES), ma anche nell’artrite reumatoide (RA). Durante la valutazione clinica reumatologica, la persona interessata deve sempre essere interrogata sulla possibile presenza di una limitazione respiratoria. La presenza di ILD nei pazienti con artrite reumatoide limita la prognosi, la sopravvivenza e la qualità di vita [1]. La diagnosi precoce è di grande importanza, in modo da poter iniziare il trattamento in tempo e prevenire una progressione rilevante dal punto di vista prognostico. La progressione dell’ILD è comune e viene osservata in misura rilevante, soprattutto nei pazienti affetti da SSc e RA; recentemente sono stati pubblicati i criteri per determinare la progressione [2]. Le esacerbazioni acute dell’ILD possono complicare ulteriormente il decorso. Queste sono associate alla progressione della malattia o sono innescate secondariamente dalle infezioni.
Consiglio direttivo ILD
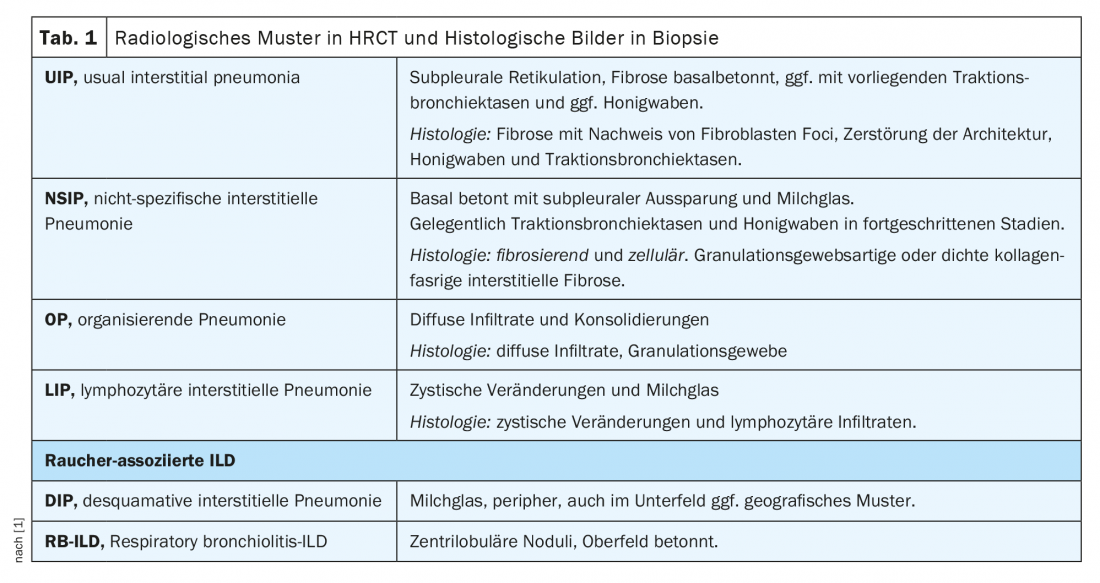
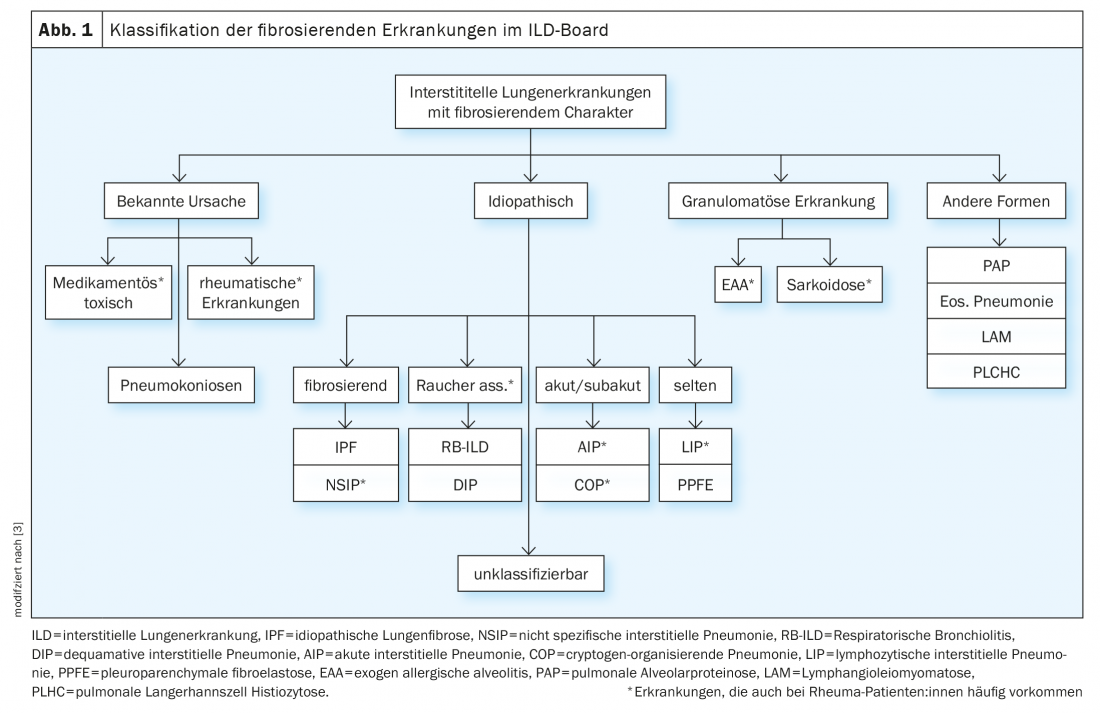
Nell’ambito della commissione ILD, i casi vengono discussi in modo interdisciplinare sulla base dei seguenti parametri: sintomi principali, anamnesi inclusa. Esposizioni e noxae, sierologia immunitaria, limitazione funzionale polmonare nella pletismografia corporea (in particolare. FVC, TLC, FEV1) e la capacità di diffusione (DLCO), la diagnostica per immagini mediante tomografia computerizzata ad alta risoluzione del torace (HRCT), nonché i risultati della diagnostica invasiva mediante broncoscopia, inclusa. Microbiologia, risultati del lavaggio broncoalveolare (BAL) e risultati istologici delle biopsie. (Tab.1). Tuttavia, i pazienti reumatici possono sviluppare l’ILD non solo come conseguenza della malattia reumatica, ma anche a causa di sostanze nocive, farmaci o esposizione (soprattutto nel caso di BAL linfocitario), ecc. (Fig. 1). Va notato che l’ottenimento di biopsie è raramente strettamente indicato nelle malattie reumatiche confermate, ma serve a semplificare la differenziazione delle diagnosi differenziali, soprattutto nelle vasculiti e soprattutto nella coesistenza di malattie tumorali e di altro tipo.
Malattie reumatiche infiammatorie con possibile coinvolgimento di ILD
RA-ILD: la RA è la malattia reumatica più comune. La presenza di artrite, soprattutto nelle piccole articolazioni, nonché di fattori reumatoidi elevati e di anticorpi contro i peptidi ciclici citrullinati (la cosiddetta sieropositività) semplificano la diagnosi e sono di conseguenza indicativi [4]. La manifestazione extra-articolare più comune della RA è l’ILD, che si riscontra nel 25-60% dei pazienti con RA alla HRCT ed è responsabile del 10-20% dei decessi per RA. La RA-ILD diventa clinicamente rilevante in circa il 10% delle persone colpite. Nel 10% dei casi, il coinvolgimento polmonare precede addirittura lo sviluppo dell’artrite [5]. I fattori di rischio per lo sviluppo di ILD includono il sesso maschile, il fumo e la sieropositività. La diagnostica per immagini mostra molto spesso un modello UIP. In letteratura sono descritte mutazioni come quelle nel promotore del gene MUC5B. Il fumo può inoltre provocare un aggravamento della situazione polmonare. La presenza di una fibrosi polmonare e di un enfisema combinati (CPFE) è associata a una prognosi drammaticamente peggiore. In modo differenziato, si deve considerare l’ILD farmaco-tossica in condizioni di immunosoppressione. Tuttavia, l’ILD associata al metotrexate (MTX) è estremamente rara (stimata allo 0,1%); è stato persino descritto un effetto protettivo del MTX.
Pertanto, il MTX non deve essere interrotto nella RA-ILD. La RA-ILD viene trattata principalmente ottimizzando l’immunosoppressione. Mancano studi controllati randomizzati per il trattamento della RA-ILD. Le migliori evidenze supportano il trattamento dei pazienti con RA e ILD concomitante con abatacept e rituximab [6]. I risultati dello studio APRIL (abatacept in RA-ILD) sono ancora in attesa. Inoltre, l’efficacia degli inibitori della JAK o dei bloccanti dell’IL-6 nella RA-ILD è stata recentemente riportata in serie di casi [7]. Nei decorsi progressivi (panoramica 1), si raccomanda l’inizio di una terapia antifibrotica. Nintedanib è approvato a questo scopo nell’UE in base ai risultati dello studio INBUILD [8]. La prognosi dei pazienti con RA-ILD è significativamente limitata nel complesso, con un tempo di sopravvivenza mediano di circa tre anni [1].

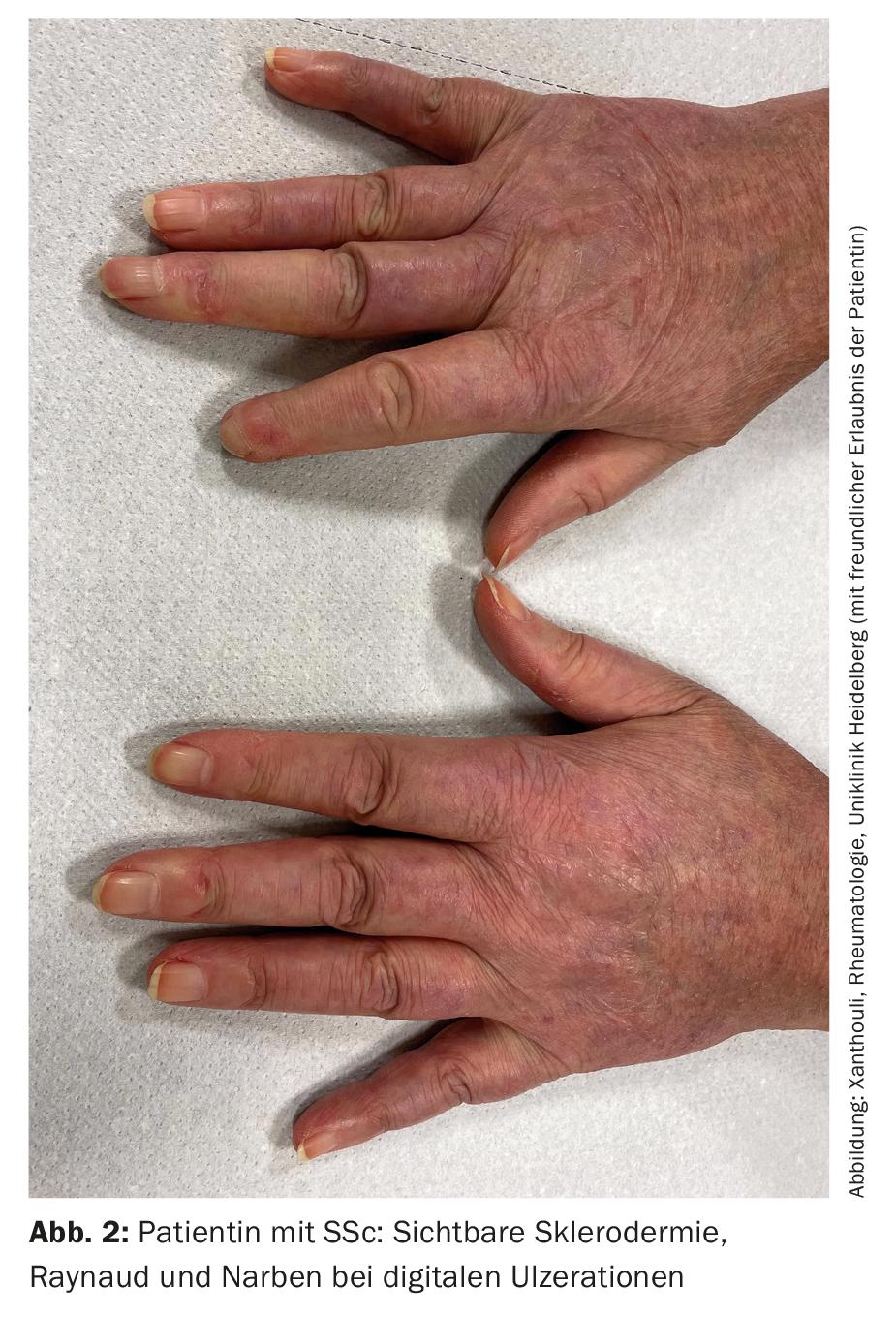
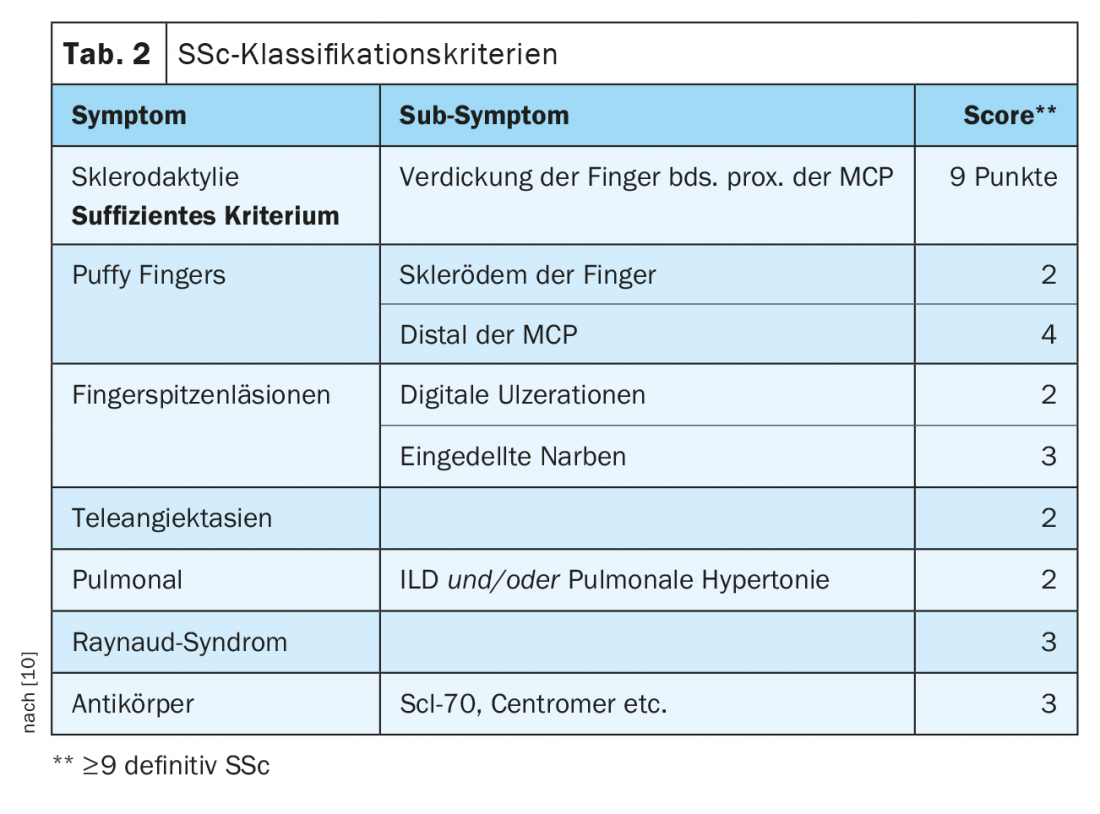
SSc-ILD: circa il 50-85% dei pazienti con SSc presenta già alterazioni polmonari interstiziali alla diagnosi iniziale. Nel 20-30% dei casi, questo è progressivo nel corso della malattia. L’ILD è attualmente la causa più comune di morte nei pazienti con SSc, con una mortalità a 10 anni di circa il 40% [1]. La diagnosi e il riconoscimento precoci e corretti dell’ILD sono prognosticamente rilevanti [9]. Sono stati stabiliti dei criteri di classificazione per semplificare la diagnosi (Tab. 2) [10]. Sclerodattilia, ulcerazioni digitali, sintomi di Raynaud e microstomia sono segni clinici che dovrebbero far pensare immediatamente alla SSc (Fig. 2). Lo sviluppo e soprattutto la progressione della malattia si verificano prevalentemente durante i primi tre anni della malattia. Spesso l’ILD è asintomatica nella SSc. Si raccomanda di iniziare lo screening con HRCT al momento della diagnosi iniziale di SSc. I test di funzionalità polmonare (pletismografia corporea e misurazione della capacità di diffusione) sono necessari ogni trimestre o semestre. I fattori di rischio per lo sviluppo di ILD sono il sesso maschile, la presenza di sclerosi sistemica cutanea diffusa, il rilevamento di autoanticorpi contro Scl-70 e l’origine afro-americana [11]. Il reflusso, l’interessamento cutaneo diffuso (e quindi un punteggio Rodnan Skin Score modificato più alto) e il sesso maschile sono fattori di rischio per una rapida progressione dell’ILD. Nella TAC, il modello NSIP è più frequentemente presente, mentre il modello UIP si trova meno frequentemente.
Il trattamento è principalmente immunomodulante con un complemento antifibrotico [1]. In base ai risultati degli studi SLS-I e -II, la terapia con ciclofosfamide o micofenolato mofetile può essere iniziata nella SSc-ILD [12,13]. Nello studio FocuSSed, la FVC è migliorata nel fenotipo SSc infiammatorio con tocilizumab (anticorpo contro il recettore IL-6). Tocilizumab ha quindi ricevuto l’approvazione della FDA per l’indicazione SSc-ILD, ma è ancora utilizzato off label in Europa [14]. Per quanto riguarda il rituximab (anch’esso ancora off label), sono attualmente disponibili buoni dati per il trattamento della SSc-ILD dallo studio DESIRES [15]; i risultati finali dello studio RECITAL sono ancora in attesa. Lo studio SCENCIS è il primo, più grande studio randomizzato, in doppio cieco e controllato per valutare l’efficacia della terapia antifibrotica con nintedanib nei pazienti con SSc-ILD [16]. Nintedanib è approvato dalla FDA per il trattamento della SSc-ILD, ma funziona solo per prevenire la progressione del coinvolgimento polmonare e non è adatto al trattamento di altre manifestazioni della SSc.
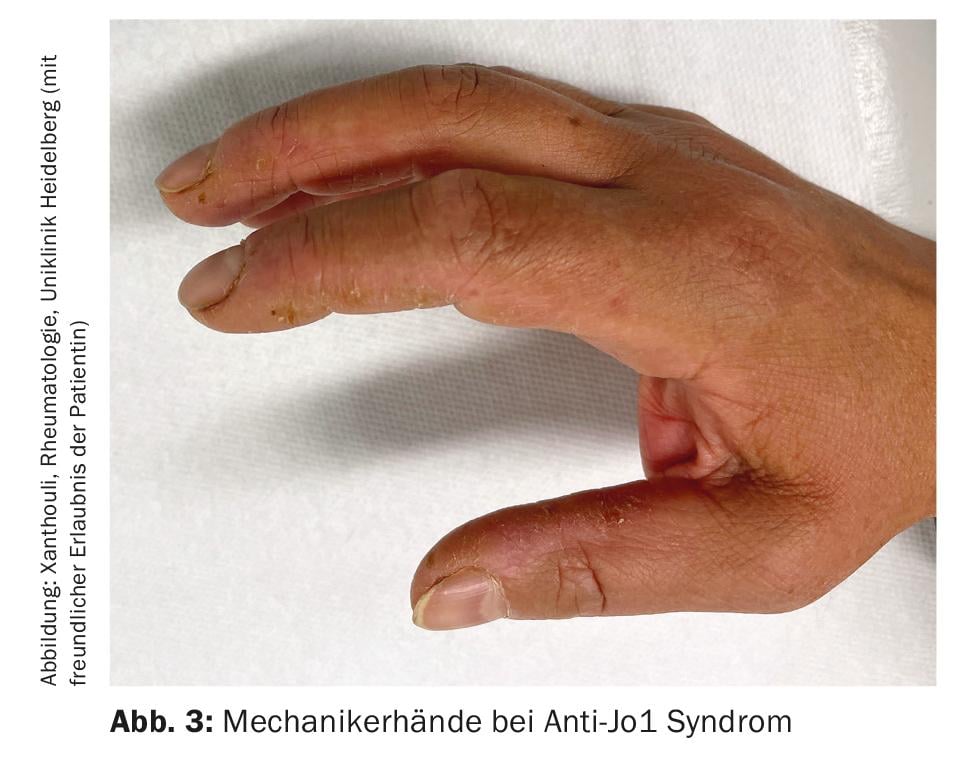
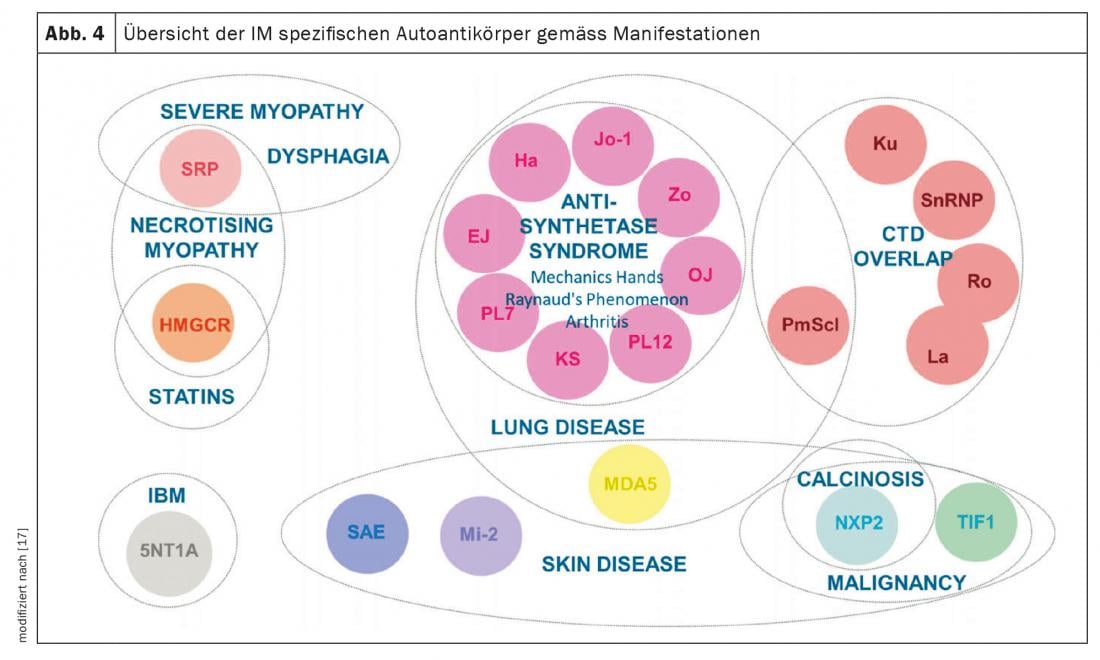
Miosite infiammatoria (IM): l ‘IM comprende la sindrome da antisintetasi (ASD), la dermato- e polimiosite, la miosite a corpi inclusi e le sindromi di sovrapposizione con altre collagenosi. Si manifestano con e senza coinvolgimento muscolare (miosite). I pazienti con coinvolgimento muscolare presentano valori muscolari eccezionalmente elevati nel sangue (CK, creatina chinasi) e mostrano segnali patologici nella risonanza magnetica muscolare e nell’esame neurofisiologico. Altre manifestazioni cliniche suggestive di IM includono le mani del meccanico (Fig. 3), soprattutto nella sindrome antisintetasi, le artriti, l’esantema eliotropico nella dermatomiosite e il fenomeno di Raynaud. Fino al 70% dei pazienti con ASA sviluppa ILD. Inoltre, l’ILD è la prima manifestazione della malattia in un terzo delle persone con IM [7]. Gli autoanticorpi più comuni nell’ASA sono gli anticorpi contro Jo-1, ma altri anticorpi, ad esempio contro PL12, PL7, Mi2, ecc. sono noti e devono essere determinati se i sintomi sono tipici. Il rilevamento di anticorpiMDA5 (melanoma differentiation accociated protein 5)in caso di coinvolgimento polmonare non chiaro indica un decorso rapidamente progressivo, refrattario alla terapia e associato a una maggiore mortalità, spesso anche senza miosite. Si noti l’associazione tra tumori maligni e IM. Questo è particolarmente elevato per il rilevamento degli anticorpi TIFγ e NXP2. In questo caso, una malignità è presente nel 30% dei casi, motivo per cui è indispensabile uno screening approfondito del tumore (Fig. 4) [17].
Il trattamento è principalmente con steroidi ad alte dosi, con una buona risposta soprattutto nei pazienti più giovani, in quelli con intervento chirurgico, livelli elevati di CK e infiltrati a vetro di latte sulla HRCT. Nei casi più gravi, si deve prendere in considerazione l’inizio di una terapia a impulsi con ciclofosfamide. Il trattamento con micofenolato, rituximab, tacrolimus, abatacept e inibitori della JAK rimane off label, anche se è supportato da buoni risultati di studi retrospettivi [18]. Vale anche la pena di notare che esiste un’autorizzazione all’immissione in commercio per la somministrazione di immunoglobuline per via endovenosa nella miosite nell’UE [19]. I pazienti con anticorpi Jo-1 ed evidenza di anticorpi anti-Ro-52 hanno recentemente mostrato una buona risposta al rituximab per quanto riguarda il coinvolgimento dell’ILD, motivo per cui dovrebbe essere considerato come opzione di trattamento [20].
LES: le multiformi manifestazioni polmonari del LES spesso portano a una diagnosi ritardata. Fino al 12% delle persone colpite potrebbe sviluppare l’ILD nel corso della malattia. Una manifestazione particolarmente pericolosa del LES è la sindrome emorragica polmonare con grave emottisi e rapido deterioramento respiratorio. D’altra parte, una dispnea grave, un disturbo restrittivo della ventilazione con aprassia diaframmatica senza la presenza di ILD sollevano il sospetto della presenza di una sindrome polmonare da contrazione, difficile da trattare. Possono essere utili un’ecografia toracica, la misurazione della forza muscolare respiratoria e la spiroergometria. Il trattamento dell’ILD nel LES consiste principalmente nell’intensificare l’immunosoppressione. I farmaci antifibrotici potrebbero essere utilizzati in caso di progressione – in casi rari; un’autorizzazione all’immissione in commercio per questo è disponibile per nintedanib nell’UE [21].
Sindrome di Sjögren: sebbene la maggior parte dei pazienti affetti da Sjögren non esprima quasi alcun sintomo polmonare, in alcuni casi si riscontra un disturbo restrittivo della ventilazione nella diagnosi della funzionalità polmonare. La presenza di sintomi sicci oggettivi con anticorpi SSA positivi è decisiva per la diagnosi della malattia di base [22]. Nel 15% dei casi, la HRCT mostra un modello LIP con alterazioni cistiche diffuse e bipolmonari con vetro di latte. La biopsia mostra un infiltrato linfocitario. In caso di masse intratoraciche poco chiare nella sindrome di Sjögren primaria, il linfoma deve essere considerato come diagnosi differenziale. La diagnosi precoce dell’ILD associata alla malattia di Sjögren è prognosticamente rilevante, con una sopravvivenza a 5 anni dell’88,5% [23].
Polmonite interstiziale con caratteristiche autoimmuni (IPAF): i pazienti affetti da ILD con anticorpi antinucleari anormali (ANA) si presentano spesso ai reumatologi. Nel 10-20% dei casi, si tratta di risultati incidentali nel corso della diagnostica di screening dell’ILD. Nella HRCT, il modello NSIP è il più comune e i soggetti colpiti traggono un beneficio significativo dagli steroidi. Tuttavia, raramente sono presenti altri sintomi indicativi di una malattia infiammatoria e reumatica. In questo caso, è necessaria un’assistenza combinata con uno pneumologo e un reumatologo per identificare in tempo una manifestazione tardiva di una malattia reumatica infiammatoria [7].
Altre malattie
Vasculitidi: Nei pazienti con granulomatosi con poliangite (GPA), spesso si possono rilevare granumolomi intrapolmonari. L’ILD può svilupparsi in casi rari. Al contrario, quando vengono rilevati gli anticorpi contro la mieloperossidasi (MPO-ANCA), viene spesso descritta una ILD con un modello prevalentemente UIP. In questo caso, è necessario fare una diagnosi complementare per quanto riguarda le manifestazioni di altri organi.
Collagenosi mista: l’incidenza di ILD è stimata molto variabile al 47-78%. I soggetti affetti presentano ANA e anticorpi U1 RNP positivi. Nella HRCT, possono essere presenti un pattern NSIP, un pattern UIP o infiltrati nel senso di un OP. Il trattamento è principalmente con sostanze immunomodulanti [7].
Altri trattamenti non medicinali e profilattici
Tutti i pazienti con malattie reumatiche dovrebbero ricevere le vaccinazioni raccomandate, soprattutto contro COVID-19, influenza e pneumococchi, e aggiornarle regolarmente. Negli stadi avanzati dell’ILD, può verificarsi un’insufficienza respiratoria. La sostituzione dell’ossigeno è spesso necessaria durante il decorso, a partire dall’esercizio fisico ed eventualmente in seguito a riposo. A questo scopo, di solito si effettua una titolazione per determinare la dose. Le misure riabilitative, come gli sport polmonari e reumatici, nonché le misure di riabilitazione in regime di ricovero o ambulatoriale, migliorano la situazione clinica e l’assorbimento di ossigeno delle persone affette da malattie reumatiche infiammatorie e sono tuttora raccomandate. Se le opzioni terapeutiche sono esaurite, si deve prendere in considerazione l’inserimento in lista per il trapianto di polmone. Non ci sono linee guida chiare sul momento esatto della presentazione per la quotazione. Nella SSc, il trapianto di cellule staminali autologhe sta diventando sempre più importante, soprattutto nei casi di decorso clinico grave [24]. Se il trapianto non è un’opzione, si deve prendere in considerazione l’avvio di misure palliative e di alleviamento dei sintomi [25].
Sommario
Le malattie reumatiche possono manifestarsi nel parenchima polmonare e portare allo sviluppo della malattia polmonare interstiziale (ILD), tra le altre cose. Le manifestazioni polmonari possono precedere la malattia reumatica sottostante in circa il 10% dei casi. Pertanto, i reumatologi sono coinvolti nel comitato interdisciplinare per le ILD per contribuire alla diagnosi differenziale e alla determinazione dell’algoritmo di trattamento. La prognosi della IOPD reumatoide è limitata, ma la collaborazione tra reumatologi e pneumologi può migliorare significativamente la qualità dell’assistenza. Sebbene la patogenesi dello sviluppo dell’ILD rimanga poco chiara, lo sviluppo di terapie farmacologiche per l’ILD reumatoide sta progredendo. Sia il reumatologo che il pneumologo devono avere una solida conoscenza e fiducia nell’uso di sostanze immunomodulanti e antifibrotiche.
Messaggi da portare a casa
- Le malattie reumatiche infiammatorie possono manifestarsi nel parenchima polmonare.
- Per la diagnosi e il trattamento, è necessaria una stretta collaborazione tra pneumologi e reumatologi.
- Il trattamento è principalmente immunosoppressivo.
- Dal 2020, la sostanza antifibrotica nintedanib è stata approvata per il trattamento della SSc-ILD in base ai risultati dello studio SENSCIS. Il farmaco viene utilizzato anche per il trattamento dell’ILD progressiva (studio INBUILD).
Letteratura:
- Wijsenbeek M, Cottin V: Spettro delle malattie polmonari fibrotiche. N Engl J Med 2020; 383: 958-968; doi: 10.1056/NEJMra2005230.
- Raghu G, Remy-Jardin M, Richeldi L et al: Fibrosi polmonare idiopatica (un aggiornamento) e fibrosi polmonare progressiva negli adulti: una linea guida ufficiale di pratica clinica ATS/ERS/JRS/ALAT. Am J Respir Crit Care Med 2022; 205: e18-e47; doi: 10.1164/rccm.202202-0399ST.
- Kreuter M, Ladner UM, Costabel U, et al.: Diagnosi e trattamento della fibrosi polmonare. Dtsch Arztebl Int 2021; 118; doi: 10.3238/arztebl.m2021.0018.
- Aletaha D, Neogi T, Silman AJ, et al: Criteri di classificazione dell’artrite reumatoide 2010: un’iniziativa collaborativa American College of Rheumatology/European League Against Rheumatism. Ann Rheum Dis 2010; 69: 1580-1588; doi: 10.1136/ard.2010.138461.
- Duarte AC, Porter JC, Leandro MJ: Il polmone in una coorte di pazienti affetti da artrite reumatoide – una panoramica dei diversi tipi di coinvolgimento e di trattamento. Rheumatology (Oxford) 2019; 58: 2031-2038; doi: 10.1093/rheumatology/kez177.
- Mena-Vázquez N, Rojas-Gimenez M, Romero-Barco CM, et al: Predittori di progressione e mortalità nei pazienti con artrite reumatoide prevalente e malattia polmonare interstiziale: uno studio prospettico di coorte. J Clin Med 2021; 10(4): 874.
- Bastian HKA: Polmone – Malattie polmonari interstiziali. Act Rheumatol 2021; 46: 544-551.
- Flaherty KR, Wells AU, Cottin V, et al: Nintedanib nelle malattie interstiziali fibrose progressive del polmone. N Engl J Med 2019; 381: 1718-1727; doi: 10.1056/NEJMoa1908681.
- Xanthouli P, Hermann W, Hunzelmann N, et al: Malattia polmonare interstiziale associata alla sclerodermia. Lo Pneumologo 2018; 15: 383-395.
- van den Hoogen F, Khanna D, Fransen J, et al: Criteri di classificazione 2013 per la sclerosi sistemica: un’iniziativa collaborativa American college of rheumatology/European league against rheumatism. Ann Rheum Dis 2013; 72: 1747-1755; doi: 10.1136/annrheumdis-2013-204424.
- Distler O, Volkmann ER, Hoffmann-Vold AM, et al: Prospettive attuali e future sulla gestione della malattia polmonare interstiziale associata alla sclerosi sistemica. Expert Rev Clin Immunol 2019; 15: 1009-1017; doi: 10.1080/1744666X.2020.1668269.
- Tashkin DP, Elashoff R, Clements PJ, et al: Ciclofosfamide rispetto al placebo nella malattia polmonare della sclerodermia. N Engl J Med 2006; 354: 2655-2666; doi: 10.1056/NEJMoa055120.
- Tashkin DP, Roth MD, Clements PJ, et al: Micofenolato mofetile rispetto alla ciclofosfamide orale nella malattia polmonare interstiziale correlata alla sclerodermia (SLS II): uno studio randomizzato controllato, in doppio cieco, a gruppi paralleli. Lancet Respir Med 2016; 4: 708-719; doi: 10.1016/S2213-2600(16)30152-7.
- Khanna D, Lin CJF, Furst DE, et al: Tocilizumab nella sclerosi sistemica: uno studio di fase 3 randomizzato, in doppio cieco, controllato con placebo. Lancet Respir Med 2020; 8(10): 963-974.
- Ebata S, Yoshizaki A, Oba K, et al: Sicurezza ed efficacia del rituximab nella sclerosi sistemica (DESIRES): uno studio in doppio cieco, avviato dallo sperimentatore, randomizzato e controllato con placebo. The Lancet Rheumatology 2021; 3: E489-E497; doi: 10.1016/S2665-9913(21)00107-7.
- Distler O, Highland KB, Gahlemann M, et al: Nintedanib per la malattia polmonare interstiziale associata alla sclerosi sistemica. N Engl J Med 2019; 380(26): 2518-2528.
- Betteridge Z, McHugh N: Autoanticorpi specifici per la miosite: uno strumento importante per supportare la diagnosi di miosite. J Intern Med 2016; 280: 8-23; doi: 10.1111/joim.12451.
- Mehta P, Aggarwal R, Porter JC, et al: Gestione della malattia polmonare interstiziale (ILD) nelle sindromi da miosite: Una guida pratica per i medici. Best Pract Res Clin Rheumatol 2022; 101769; doi: 10.1016/j.berh.2022.101769.
- Aggarwal R, Charles-Schoeman C, Schessl J, et al: Studio di fase III prospettico, in doppio cieco, randomizzato, controllato con placebo, che valuta l’efficacia e la sicurezza di octagam 10% nei pazienti con dermatomiosite (“Studio ProDERM”). Medicine (Baltimora) 2021; 100: e23677; doi: 10.1097/MD.0000000000023677.
- Bauhammer J, Blank N, Max R, et al: Rituximab nel trattamento della Sindrome da Antisintetasi associata all’anticorpo Jo1: positività anti-Ro52 come marcatore di gravità e risposta al trattamento. J Rheumatol 2016; 43: 1566-1574; doi: 10.3899/jrheum.150844.
- Medlin JL, Hansen KE, McCoy SS, et al: Manifestazioni polmonari nel lupus eritematoso sistemico tardivo rispetto a quello precoce: revisione sistematica e meta-analisi. Semin Arthritis Rheum 2018; 48: 198-204; doi: 10.1016/j.semarthrit.2018.01.010.
- Shiboski CH, Shiboski SC, Seror R, et al: Criteri di classificazione dell’American College of Rheumatology/European League Against Rheumatism del 2016 per la sindrome di Sjogren primaria: un consenso e una metodologia guidata dai dati che ha coinvolto tre coorti internazionali di pazienti. Arthritis Rheumatol 2017; 69: 35-45; doi: 10.1002/art.39859.
- Gao H, Zhang XW, He J, et al: Prevalenza, fattori di rischio e prognosi della malattia polmonare interstiziale in un’ampia coorte di pazienti cinesi con sindrome di Sjogren primaria: uno studio caso-controllo. Medicina (Baltimora) 2018; 97: e11003; doi: 10.1097/MD.0000000000011003.
- Farge D, Ait Abdallah N, Marjanovic Z, et al: Trapianto autologo di cellule staminali nella sclerodermia. Press Med 2021; 50: 104065; doi: 10.1016/j.lpm.2021.104065.
- Kreuter M, Bendstrup E, Russell AM, et al: Cure palliative nella malattia polmonare interstiziale: vivere bene. Lancet Respir Med 2017; 5: 968-980; doi: 10.1016/S2213-2600(17)30383-1.
InFo PNEUMOLOGIA & ALLERGOLOGIA 2022; 4(3): 8-13









