Il termine collettivo “ipertensione polmonare (PH)” comprende diversi tipi di ipertensione polmonare pericolosi per la vita, i cui approcci terapeutici sono molto diversi e altamente complessi. Poiché i sintomi sono spesso aspecifici, anche la diagnosi è una grande sfida.
La classificazione clinica dell’ipertensione polmonare (PH) è attualmente suddivisa in cinque gruppi, come ci ha ricordato all’inizio il Prof. Dr. Horst Olschewski, Capo del Dipartimento Clinico di Pneumologia, Dipartimento Universitario di Medicina Interna, Ospedale Universitario LKH di Graz:
- Il gruppo 1 comprende l’ipertensione arteriosa polmonare (PAH), la forma più rara di ipertensione polmonare, che viene diagnosticata solo dopo aver escluso altre cause sottostanti.
- Il gruppo 2, l’ipertensione polmonare nella cardiopatia sinistra, è di gran lunga la forma più comune di ipertensione polmonare.
- Il gruppo 3 comprende l’ipertensione polmonare dovuta a malattie polmonari e/o a carenza di ossigeno. Questa forma si presenta anche molto più frequentemente dell’IPA.
- Il gruppo 4 comprende l’ipertensione polmonare a seguito di embolia polmonare, che colpisce fino al 4% dei pazienti che hanno subito un’embolia polmonare acuta.
- Il Gruppo 5 comprende forme rare di ipertensione polmonare che non possono essere chiaramente assegnate a nessuno dei gruppi sopra citati, come l’ipertensione polmonare nella sarcoidosi o nella malattia renale cronica.
I gruppi 2 e 3, secondo il Prof. Olschewski, insieme rappresentano un totale del 90% di tutta l’ipertensione polmonare, il che pone dei pericoli diagnostici differenziali.
Eterogeneità dell’IPAH
Per l’analisi dei cluster, sono stati identificati i pazienti che rispondevano ai criteri dell’ipertensione arteriosa polmonare idiopatica (IPAH).
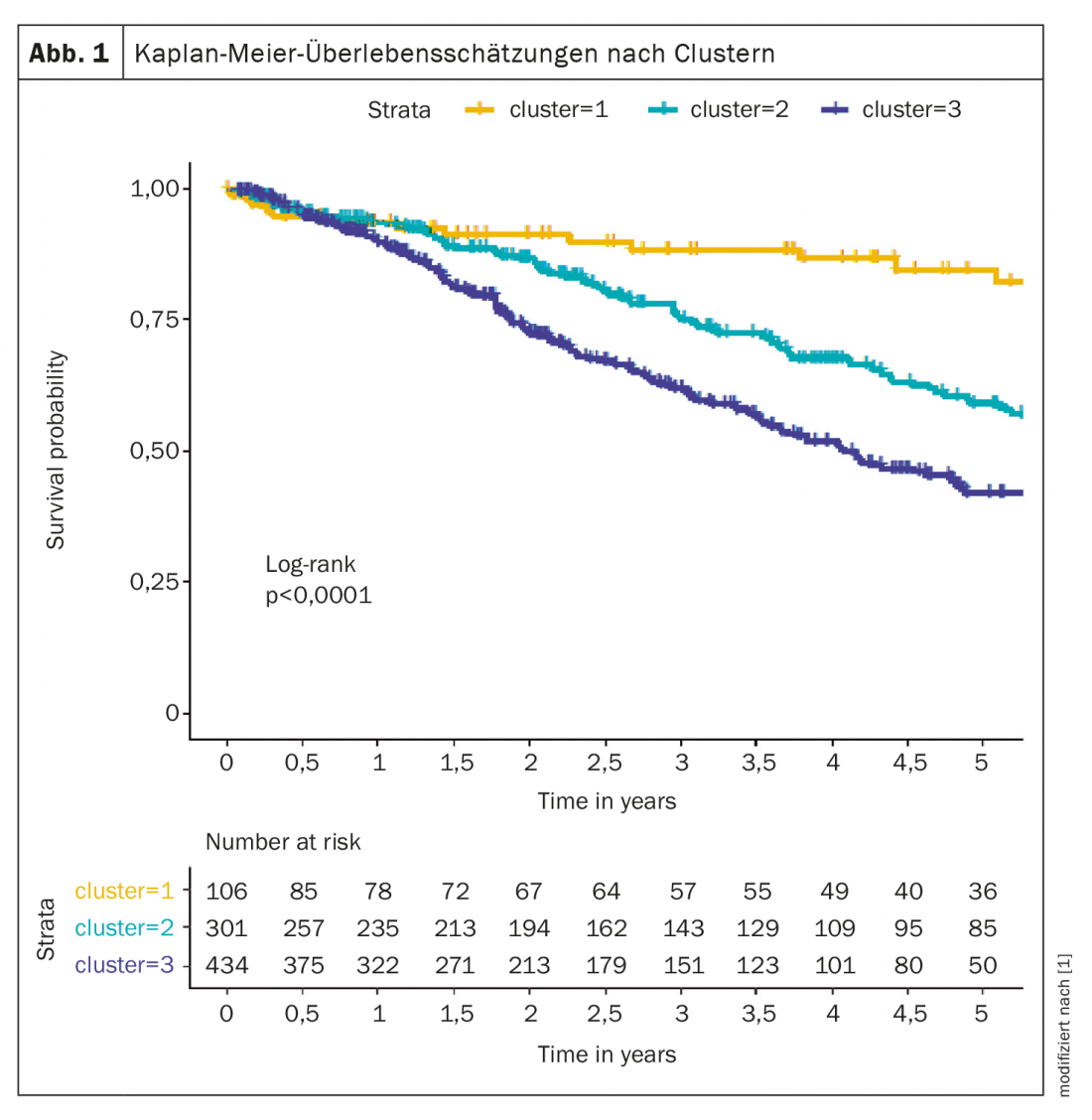
Per esempio, un’analisi dei cluster ha identificato diversi fenotipi che differiscono nella presentazione clinica, nella risposta alla terapia e nella sopravvivenza. I gruppi analizzati sono stati composti dal registro COMPERA [1], sono stati definiti tre cluster: il cluster 1 (n=106; 12,6%) comprendeva un’età media di 45 anni, il 76% erano donne, nessuna comorbilità, per lo più mai fumatori, DLCO ≥45%; il cluster 2 (n=301; 35,8%) comprendeva pazienti la cui età media era di 75 anni, il 98% donne, frequenti comorbilità, nessuna storia di fumo, DLCO per lo più ≥45%; il cluster 3 (n=434; 51,6%) consisteva in soggetti con un’età media di 72 anni, il 72% erano uomini, frequenti comorbidità, storia di fumo e DLCO per lo più inferiore al 45%.
I pazienti del cluster 1 hanno risposto meglio al trattamento della PAH rispetto ai pazienti degli altri due cluster. Il tasso di sopravvivenza a cinque anni è stato dell’84,6% nel cluster 1, del 59,2% nel cluster 2 e del 42,2% nel cluster 3 (Fig. 1). Secondo l’esperto, questi dati suggeriscono che sono necessari dei criteri per distinguere i pazienti con IPAH atipica da quelli con IPAH vera.
La diagnosi differenziale a volte è difficile
Poiché le conseguenze terapeutiche dipendono in larga misura dalla causa dell’ipertensione polmonare, è importante completare le procedure diagnostiche e determinare la causa principale della PH prima di prendere una decisione sui farmaci per la PAH. Il World Symposia on Pulmonary Hypertension (WSPH ) ha prodotto delle linee guida per queste importanti decisioni. La PH del gruppo 2 o le malattie evolutive complesse con aumento della pressione post-capillare possono essere riconosciute con relativa facilità dall’aumento delle pressioni di cuneo arterioso polmonare. Il gruppo PH 4 può essere rilevato o escluso dalla scansione polmonare di perfusione in combinazione con la TAC del torace. La PAH di gruppo 1 e la PH di gruppo 3 sono profili patologici molto diversi, ma a volte possono essere difficili da distinguere. Il WSPH suggerisce che un’ipertensione polmonare grave combinata con una lieve compromissione del test di funzionalità polmonare (FEV1 >60 e FVC >60%), lievi anomalie parenchimali alla TAC ad alta risoluzione del torace e una compromissione circolatoria al test da sforzo cardiopolmonare sono indicativi del gruppo PAH 1. Questi pazienti sono candidati alla terapia della PAH. Se il paziente soffre di PH di gruppo 3, l’unica indicazione possibile per la terapia della PAH è l’ipertensione polmonare grave (mPAP ≥35 mmHg o mPAP tra 25 e 35 mmHg insieme a un indice cardiaco (CI) molto basso <2,0 L/min/m2), che può essere ricavata solo in modo invasivo, ha detto il Prof. Olschewski.
Uno studio ha anche esaminato la differenziazione tra i pazienti con PAH (gruppo 1) e i pazienti con insufficienza cardiaca (gruppo 2) [2]. Sebbene l’aumento delle pressioni di riempimento del lato sinistro e il rigurgito mitralico funzionale siano le cause principali della PH postcapillare, le linee guida e le raccomandazioni distinguono tra PH postcapillare isolata (IpcPH) e PH combinata post e precapillare (CpcPH). Quest’ultima è definita dalla resistenza vascolare polmonare (PVR) aumentata alle unità di Wood (WE). È importante distinguere tra la definizione generale di PH (mPAP >20 mmHg) e la definizione di PH precapillare che include la PAH, per la quale è richiesta anche una pressione di cuneo arterioso polmonare (PAWP) ≤15 mmHg e un aumento della resistenza vascolare polmonare (PVR) a ≥3 unità Wood. Secondo lo studio, la terapia farmacologica mirata per la PAH è indicata per una PAWP ≤15 mmHg.
Anche la differenziazione dei pazienti con PAH e BPCO (gruppo 1) dai pazienti con PH dovuta a BPCO (gruppo 3) non è facile. La maggior parte dei pazienti con BPCO con PH appartiene al gruppo 3. Alcuni pazienti con BPCO con PH e aumento della pressione di riempimento del ventricolo sinistro (PH postcapillare) causata da una malattia cardiovascolare concomitante sono assegnati al gruppo 2. Anche le malattie tromboemboliche croniche possono causare la PH, soprattutto perché la BPCO è un fattore di rischio per il tromboembolismo venoso, questi pazienti con BPCO sono solitamente assegnati al gruppo 4. I pazienti con un’ostruzione delle vie aeree periferiche molto lieve e una grave PH precapillare con un forte aumento della resistenza vascolare polmonare (PVR) e una bassa gittata cardiaca (CO) sono prevalentemente ritenuti affetti da PAH (gruppo 1) con una lieve BPCO come condizione associata. Nella maggior parte dei casi, la PH è relativamente lieve nei pazienti con BPCO, ha spiegato il pneumologo, ma in un sottogruppo di pazienti con BPCO, la presenza di alcune caratteristiche cliniche suggerisce un “fenotipo vascolare polmonare”. Un tale fenotipo sarebbe caratterizzato da una grave PH precapillare con resistenza vascolare polmonare marcatamente aumentata, moderata limitazione del flusso d’aria, capacità diffusiva gravemente ridotta per il monossido di carbonio, normo- o ipocapnia, limitazione del carico circolatorio e insufficienza cardiaca destra progressiva.
Un altro studio ha cercato di determinare soglie emodinamiche prognosticamente rilevanti per la PH grave nella BPCO, utilizzando un approccio imparziale [3]. Un PVR >5 WU è risultato essere il più forte predittore emodinamico indipendente di mortalità nei pazienti con BPCO. Questa soglia è migliore per identificare i pazienti con BPCO e malattia vascolare polmonare grave.
I progressi nella terapia farmacologica
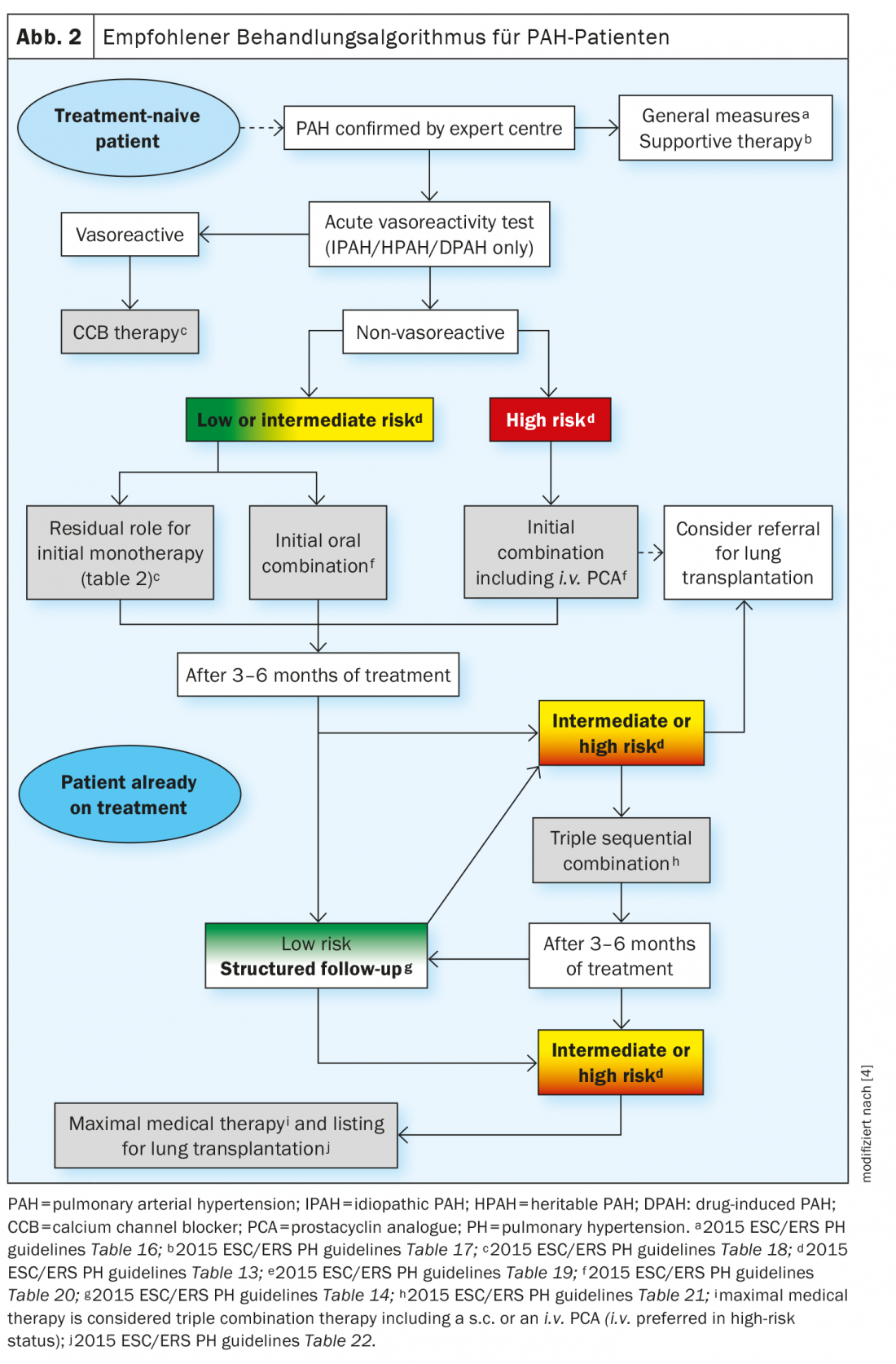
I recenti progressi nella terapia farmacologica per la PAH non sono dovuti alla scoperta di nuove vie di segnalazione, ma allo sviluppo di nuove strategie per la terapia combinata e all’escalation dei trattamenti basata sulla valutazione sistematica della risposta clinica. La strategia di trattamento si basa sulla gravità del paziente con PAH di nuova diagnosi, che viene determinata utilizzando un approccio di stratificazione del rischio multiparametrico. I parametri clinici, di esercizio, di funzione ventricolare destra ed emodinamici sono combinati per definire lo stato di rischio basso, intermedio o alto, in base alla mortalità attesa a 1 anno. L’attuale algoritmo di trattamento fornisce la strategia iniziale più appropriata, tra cui la monoterapia, la terapia di combinazione doppia o tripla. È necessaria un’ulteriore escalation del trattamento se lo stato di basso rischio non viene raggiunto alle visite di follow-up programmate. Nei casi più avanzati, può essere necessario il trapianto di polmone con la massima terapia medica (Fig. 2).
I farmaci approvati per la terapia orale includono gli inibitori della fosfodiesterasi 5 (PDE5i), come il sildenafil e il tadalafil, gli antagonisti del recettore dell’endotelina (ERA), come il bosentan, l’armbrisentan e il macitentan, gli stimolatori della guanilato ciclasi solubile (sGC), come il riociguat e gli agonisti del recettore della prostaciclina, come il selexipag. È anche possibile trattare la PAH con una terapia inalatoria o con infusioni continue.
Studi prospettici sulla terapia
Ad esempio, riociguat e gli inibitori della fosfodiesterasi-5 (PDE5i), approvati per il trattamento dell’ipertensione arteriosa polmonare (PAH), agiscono attraverso meccanismi diversi nello stesso percorso. Lo studio REPLACE ha quindi esaminato se riociguat possa essere un’opzione alternativa per i pazienti con PAH che non rispondono adeguatamente al trattamento con PDE5i. L’obiettivo era quello di valutare gli effetti del passaggio a riociguat dalla terapia con PDE5i rispetto al proseguimento della terapia con PDE5i nei pazienti con PAH a rischio intermedio di mortalità a 1 anno. I risultati mostrano che il passaggio dal trattamento con PDE5i a riociguat, che agiscono entrambi attraverso la via di segnalazione tra la guanilato ciclasi solubile dell’ossido nitrico e la guanosina monofosfato ciclica, può essere un’opzione strategica per l’escalation del trattamento nei pazienti con PAH a rischio intermedio di mortalità a 1 anno [5].
Sotatercept, un nuovo antagonista dell’activina, lega le activine e i fattori di differenziazione della crescita nel tentativo di ripristinare l’equilibrio tra le vie di segnalazione che promuovono e inibiscono la crescita. Nello studio PULSAR, 24 settimane di trattamento con sotatercept in pazienti con ipertensione arteriosa polmonare che stavano ricevendo una terapia di fondo per essa, hanno portato a una riduzione della resistenza vascolare polmonare [6]. Anche la performance fisica (misurata dalla distanza di cammino di 6 minuti) e i livelli di NT-proBNP sono migliorati con il sotaterapeuta, secondo il Prof. Olschewski.
“Sensation” dagli Stati Uniti
Il Prof. Olschewski ha avuto una “sensazione” da riferire dagli Stati Uniti: Mai prima d’ora era stata approvata una terapia per la PH nelle malattie polmonari (gruppo 3). Ora le cose sono cambiate, almeno negli Stati Uniti. Il treprostinil per via inalatoria è stato utilizzato per la prima volta in questo gruppo di pazienti. Lo studio INCREASE, controllato con placebo e della durata di 16 settimane, ha arruolato pazienti con malattia polmonare interstiziale e ipertensione polmonare, ai quali è stato somministrato treprostinil per via inalatoria mediante un nebulizzatore a ultrasuoni con somministrazione pulsata in un massimo di 12 respiri (72 μg in totale) quattro volte al giorno. Rispetto al placebo, il treprostinil per via inalatoria ha migliorato significativamente le prestazioni fisiche dei pazienti, misurate dal test del cammino di 6 minuti [7]. Negli Stati Uniti, il principio attivo è già stato approvato dalla FDA, ma se il produttore richiederà anche all’Agenzia Europea dei Medicinali (EMA) nel prossimo futuro o se attenderà ulteriori studi è ancora aperto, come ha spiegato l’esperto. “In ogni caso, è un barlume di speranza”.

L’addestramento al movimento standardizzato è stato stabilito con successo
Nello studio randomizzato controllato EU-TRAIN sull’allenamento all’esercizio fisico nei pazienti con ipertensione arteriosa polmonare (PAH) e ipertensione polmonare cronica tromboembolica (CTEPH), condotto in 11 centri di 10 Paesi europei su un’ampia popolazione di pazienti, è stato osservato un miglioramento significativo e clinicamente significativo dell’endpoint primario 6MGT e degli endpoint secondari WHO-FC, QoL e consumo massimo di ossigeno [8]. Lo studio ha dimostrato per la prima volta che un programma di esercizio fisico sicuro ed efficace come aggiunta alla terapia farmacologica può essere standardizzato e implementato in diversi Paesi con sistemi sanitari differenti.
Fonte: Aggiornamento Pneumo 2021: Ipertensione polmonare, 12.11.2021
Letteratura:
- Hoeper, et al: Fenotipi di ipertensione arteriosa polmonare idiopatica determinati dall’analisi dei cluster del registro COMPERA. J Heart Lung Transplant 2020, doi: 10.1016/j.healun.2020.09.011.
- Rosenkranz, et al: Ipertensione polmonare nell’HFpEF e nell’HFrEF: fisiopatologia, diagnosi, approcci terapeutici. Cuore 2019, doi: 10.1007/s00059-019-4831-6.
- Zeder, et al: L’elevata resistenza vascolare polmonare predice la mortalità nei pazienti con BPCO. Eur Respir J 2021, doi: 10.1183/13993003.00944-2021.
- Galiè, et al: Stratificazione del rischio e terapia medica dell’ipertensione arteriosa polmonare. European Respiratory Journal 2019, doi: 10.1183/13993003.01889-2018.
- Hoeper, et al: Passaggio a riociguat rispetto alla terapia di mantenimento con inibitori della fosfodiesterasi-5 nei pazienti con ipertensione arteriosa polmonare (REPLACE): uno studio multicentrico, open-label, randomizzato e controllato. Lancet Respir Med 2021, doi: 10.1016/S2213-2600(20)30532-4.
- Humbert, et al: Sotatercept per il trattamento dell’ipertensione arteriosa polmonare. N Engl J Med 2021, doi: 10.1056/NEJMoa2024277.
- Waxman, et al: Treprostinil per via inalatoria nell’ipertensione polmonare dovuta alla malattia polmonare interstiziale. N Engl J Med 2021, doi: 10.1056/NEJMoa2008470.
- Grüning, et al: L’allenamento all’esercizio fisico standardizzato è fattibile, sicuro ed efficace nell’ipertensione polmonare arteriosa e tromboembolica cronica: risultati di un ampio studio multicentrico europeo randomizzato e controllato. Eur Heart J 2021, doi: 10.1093/eurheartj/ehaa696.
InFo PNEUMOLOGIA & ALLEROLOGIA 2022; 4(1): 20-22