I sintomi aspecifici nella fase iniziale portano a una significativa latenza diagnostica di questi linfomi primari a cellule T cutanei. La fotoferesi extracorporea e le nuove terapie mirate stanno migliorando sempre più il successo terapeutico nella sindrome di Sézary.
La sindrome di Sézary è una malattia rara, spesso difficile da diagnosticare, aggressiva, appartenente al gruppo dei linfomi primari cutanei a cellule T (CTCL) [1,2]. La malattia appartiene ai linfomi non-Hodgkin extranodali ed è caratterizzata dalla triade di eritroderma pruritico, linfadenopatia generalizzata e cellule T neoplastiche con nucleo cerebriforme (cellule di Sézary) nel sangue, nella pelle e nei linfonodi [3,25]. La sindrome di Sézary si presenta con un’incidenza di 0,1/100.000 soprattutto tra il 40° e il 60° anno di età. Gli uomini e gli afroamericani sono colpiti più spesso delle donne e dei caucasici [4,5].
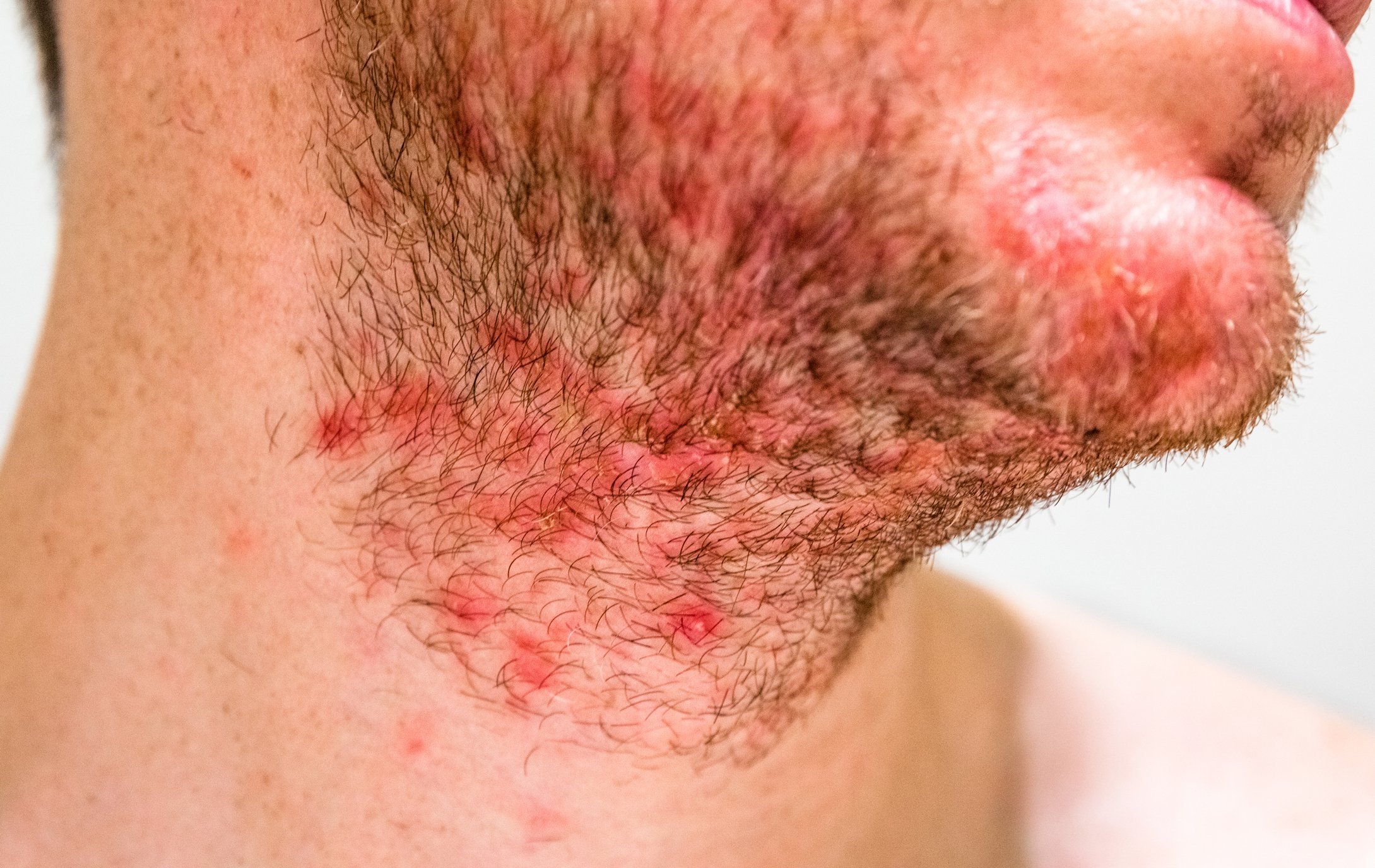
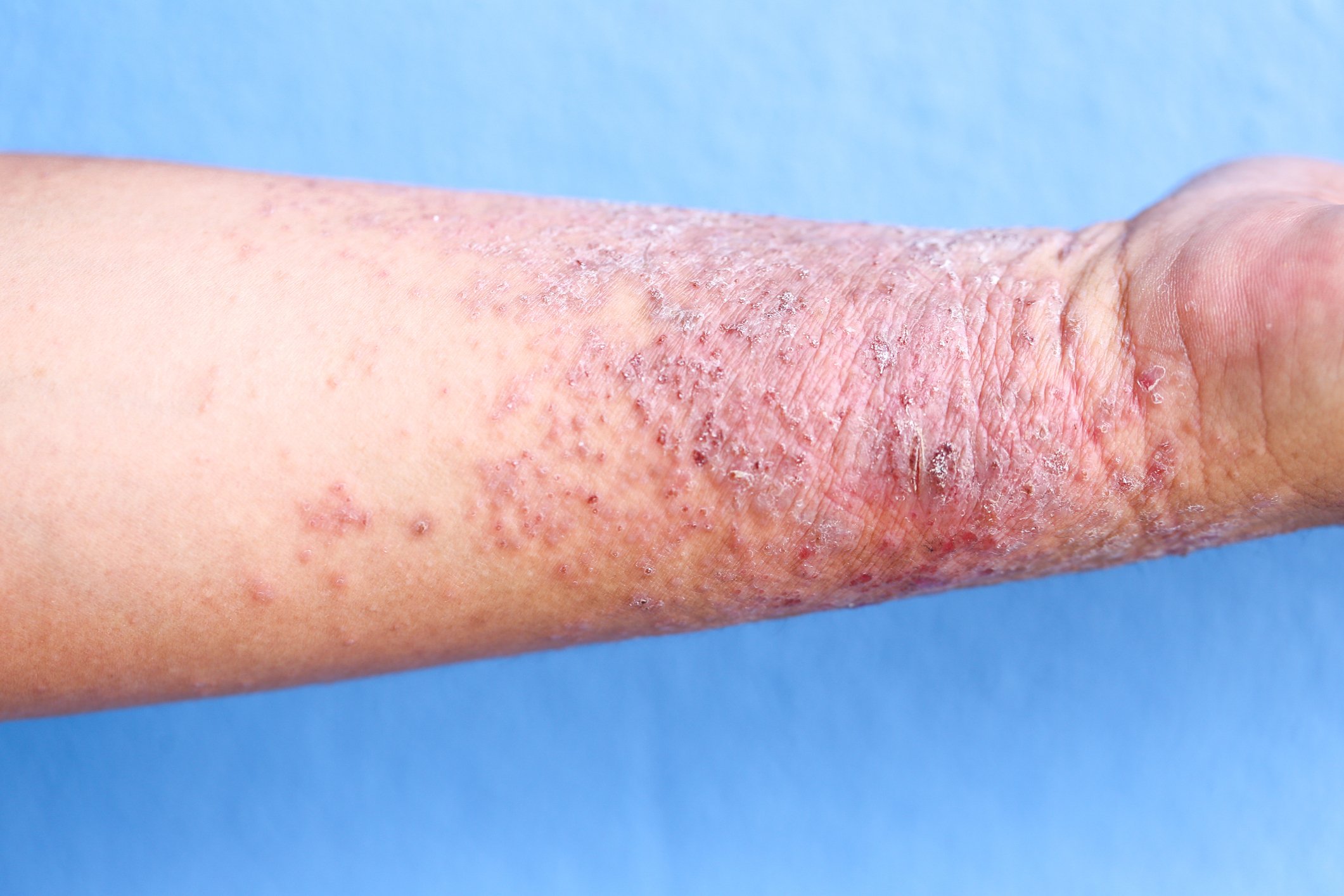
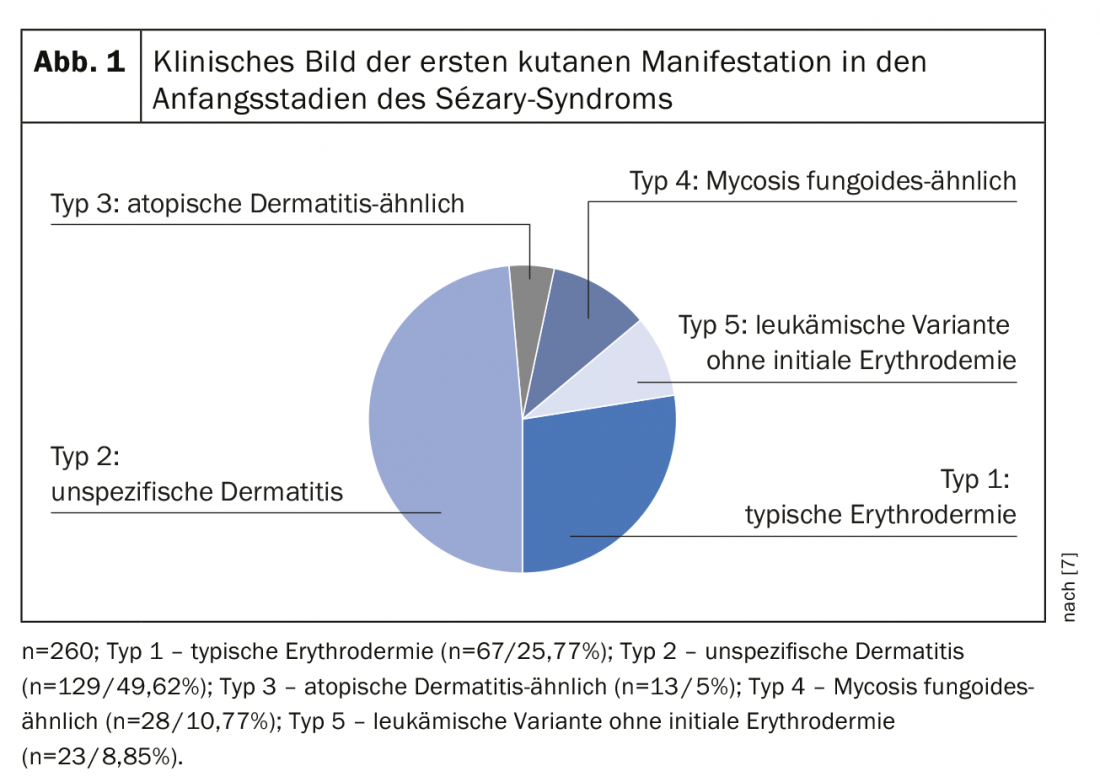
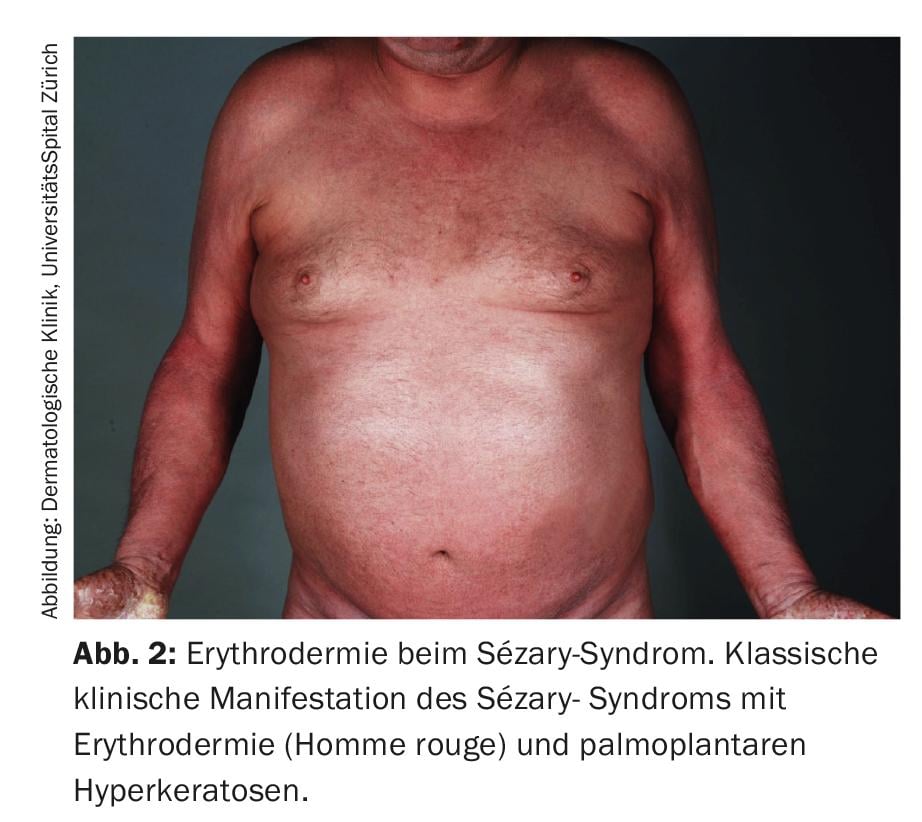
La definizione della sindrome di Sézary da parte dell’International Cutaneous Lymphoma Society insieme all’Organizzazione Mondiale della Sanità include l’eritroderma come sintomo impellente accompagnato da almeno due delle seguenti manifestazioni: >1000 cellule di Sézary/µL nel sangue periferico, anomalie immunofenotipiche delle cellule T Rapporto CD4/CD8 ≥10, cellule CD4+/CD7- ≥30%, o cellule CD4+/CD26- ≥ 40%. [25]La presenza di una popolazione di recettori monoclonali delle cellule T (TZR) nel sangue o di cloni di cellule T cromosomicamente alterati [6]. Uno studio di coorte retrospettivo pubblicato da Mangold et al. Tuttavia, ha dimostrato che solo il 25,5% dei pazienti presenta il tipico eritroderma, che colpisce >l’80% della superficie corporea, come manifestazione iniziale. Tuttavia, nell’86,3% dei pazienti, l’eritroderma si sviluppa nel tempo ed è presente al più tardi al momento della diagnosi. La presentazione clinica iniziale, spesso aspecifica, è quindi responsabile di un ritardo diagnostico compreso tra un mese e 32 anni (media 4,2 anni) (Fig. 1) [7]. Altri sintomi della sindrome di Sézary sono l’alopecia, l’onicodistrofia, l’ipercheratosi palmoplantare e il prurito massiccio, che esercita una grande sofferenza sui pazienti colpiti (Fig. 2). La perdita dell’integrità cutanea comporta un aumento del rischio di infezione da parte della flora cutanea residente, come lo Staphylococcus aureus. Gli infiltrati cutanei tumorali associati all’edema e all’ipalbuminemia possono portare alla perdita di liquidi.
Diagnosi
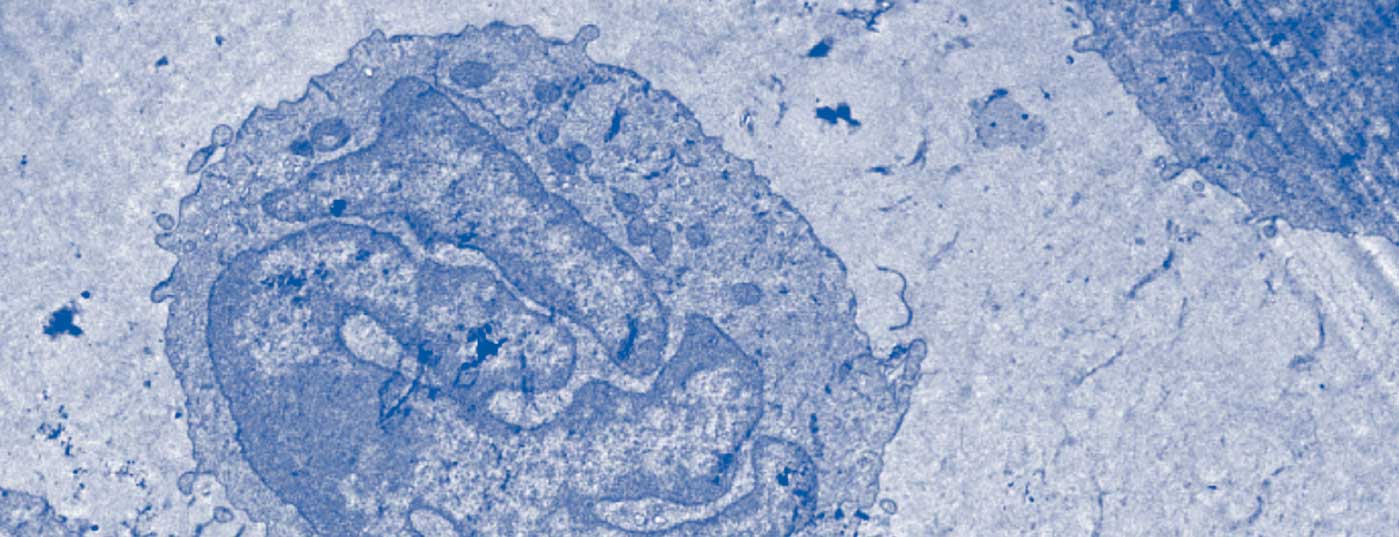
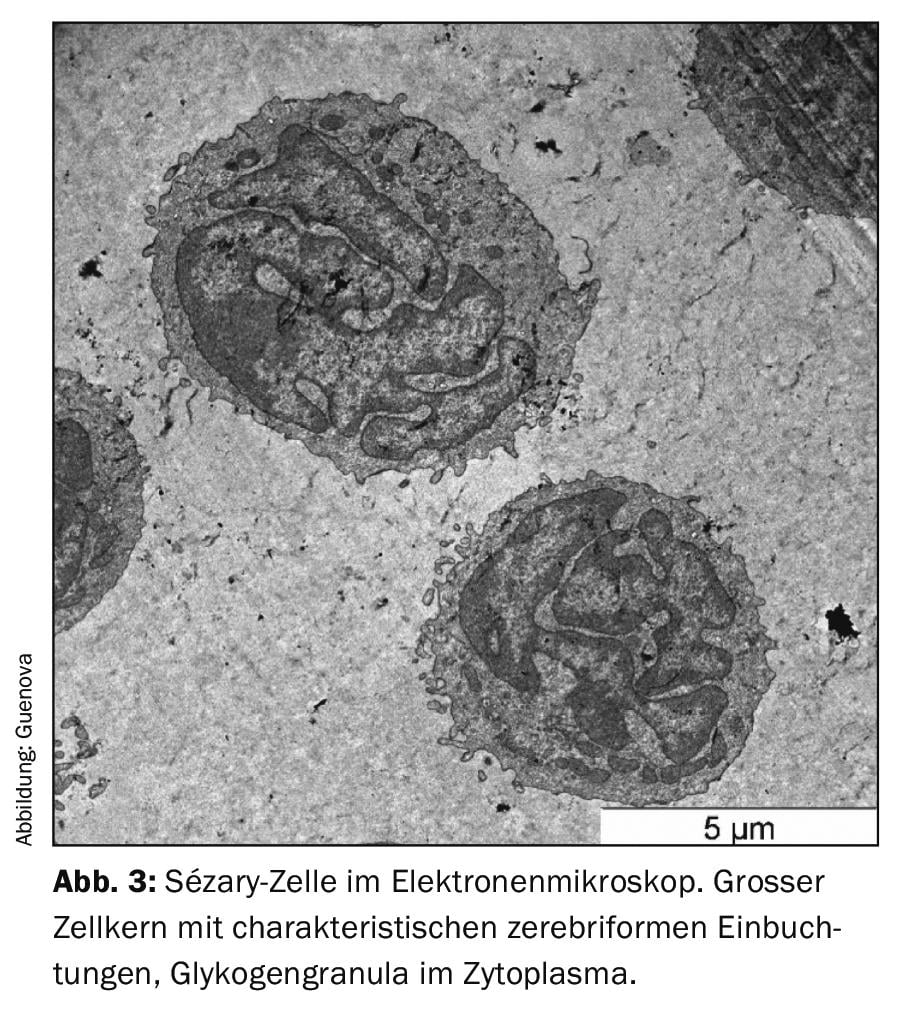
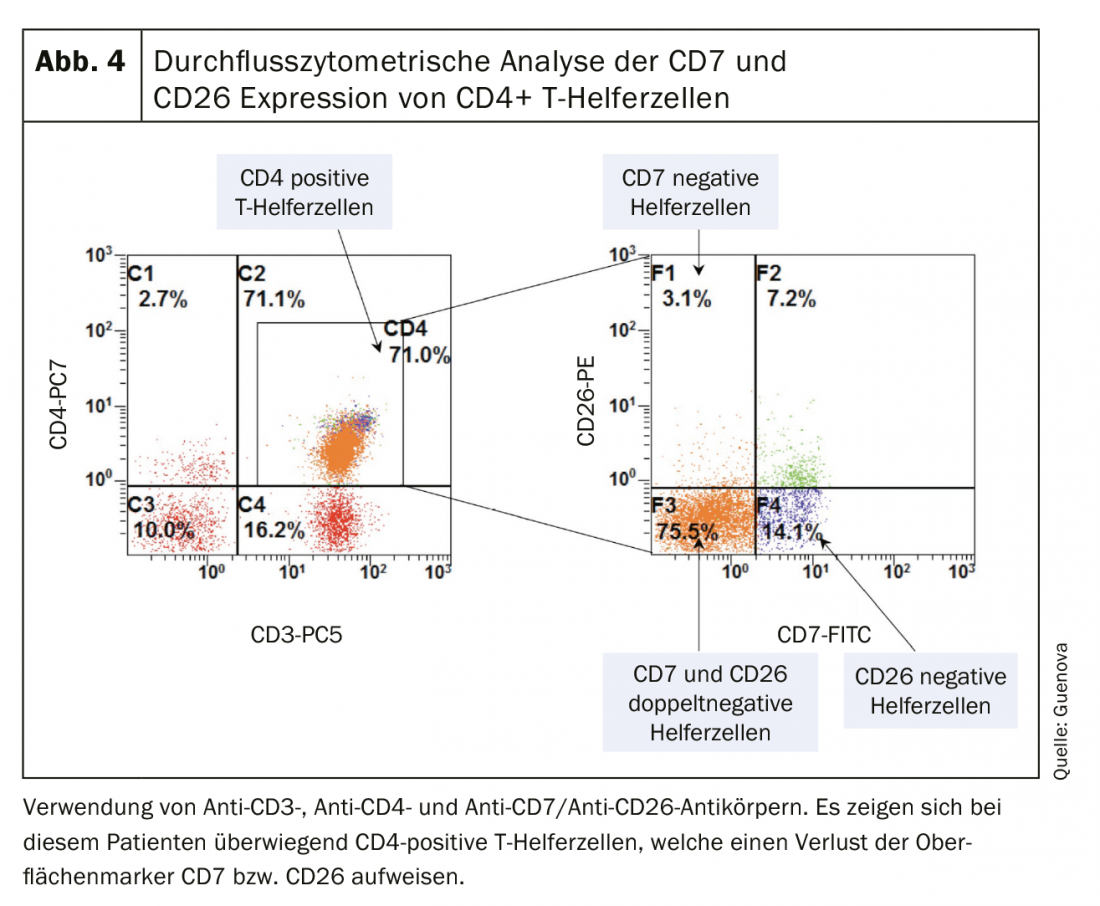
Come per molte malattie eritrodermiche della pelle, la mancanza di chiari marcatori diagnostici rende la diagnosi differenziale della sindrome di Sézary una sfida [3,8]. L’esame clinico indispensabile comprende la palpazione di tutte le regioni linfonodali, oltre a un’indagine accurata dello stato della pelle. Il cosiddetto “Modified Skin Weighted Assessment Tool” (mSWAT) [8,9] è particolarmente adatto per misurare e valutare il coinvolgimento cutaneo. Vengono esaminate biopsie multiple di lesioni cutanee rappresentative, utilizzando metodi istopatologici e immunoistochimici [3]. Istologicamente, si possono rilevare infiltrati monomorfi di cellule T atipiche, una caratteristica epidermotropia e microabscessi di Pautrier – accumuli intraepidermici di cellule maligne [8]. In circa il 40% dei casi, tuttavia, i risultati istologici non sono conclusivi per la diagnosi [10]. Il sangue periferico viene esaminato per verificare la presenza di una popolazione anormale di cellule T CD4+ e di cellule T neoplastiche morfologicamente evidenti (striscio di sangue), utilizzando la citometria a flusso e metodi molecolari o citogenetici. Queste tipiche cellule di Sézary (o anche note come cellule di Lutzner) sono caratterizzate da una morfologia ultrastrutturale con il loro nucleo cerebriforme dentellato. (Fig.3). Non solo esprimono i recettori delle chemiochine CCR7 e CCR4, ma mostrano anche una maggiore espressione di L-selectina e una caratteristica carenza degli antigeni di superficie CD7 e CD26. (Fig.4) [3,11]. Il rilevamento della clonalità dei recettori delle cellule T ha un alto valore diagnostico. [12]. La sua rilevazione biologica molecolare mediante la reazione a catena della polimerasi è stata stabilita come standard [13]. L’analisi citometrica a flusso dell’antigene Vβ del recettore delle cellule T viene utilizzata di routine nei centri universitari specializzati [12].
Recenti analisi hanno dimostrato un’aumentata espressione dell’antigene di superficie CD164 nella conta totale delle cellule T CD4+ nei pazienti con sindrome di Sézary. Questo marcatore potrebbe quindi agire come un promettente parametro diagnostico e un potenziale bersaglio terapeutico in futuro [14,15]. Altri nuovi biomarcatori ematici e cutanei, tra cui PD-1 (CD279) e KIR3DL2 (CD158k), possono aiutare a distinguere la sindrome di Sézary dalle dermatosi infiammatorie eritrodermiche [25].
Stadiazione e prognosi
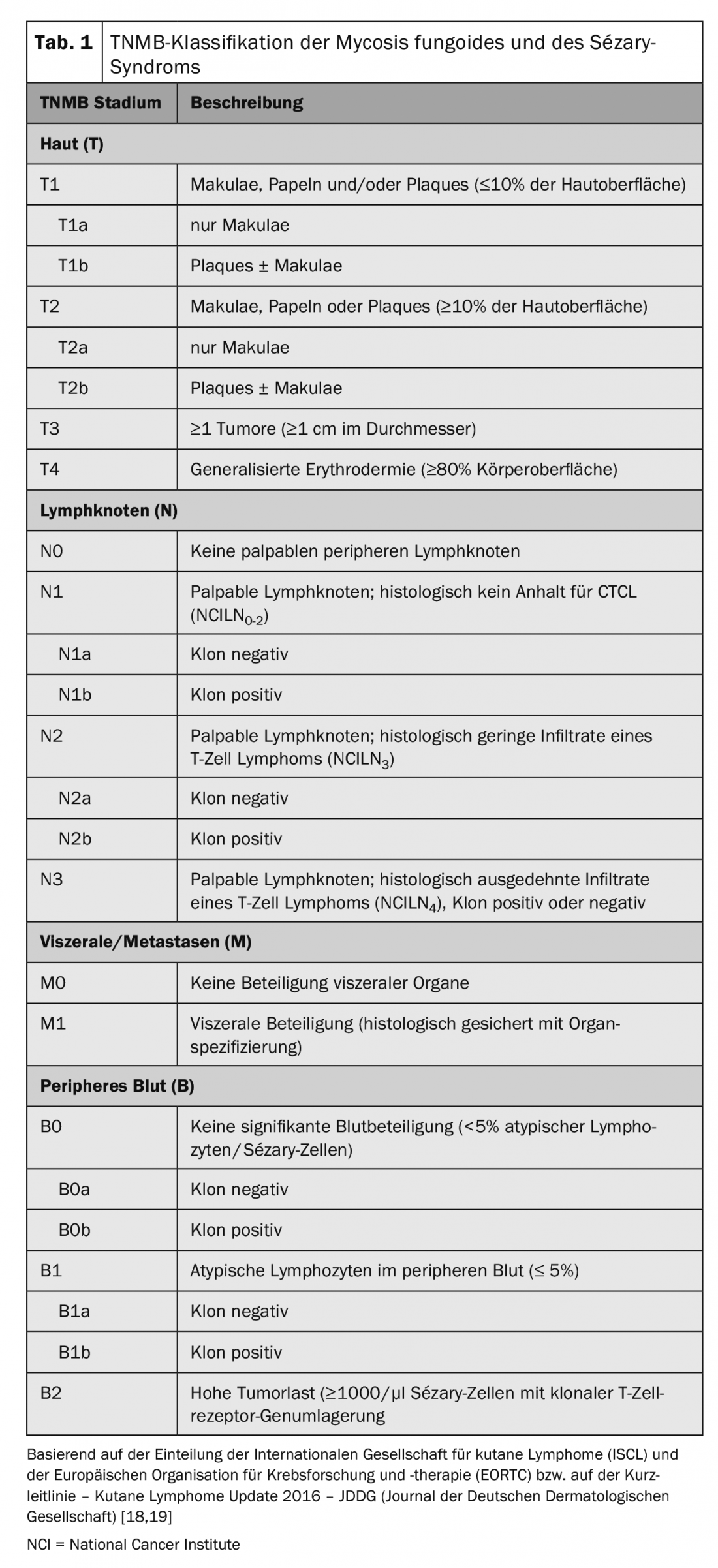
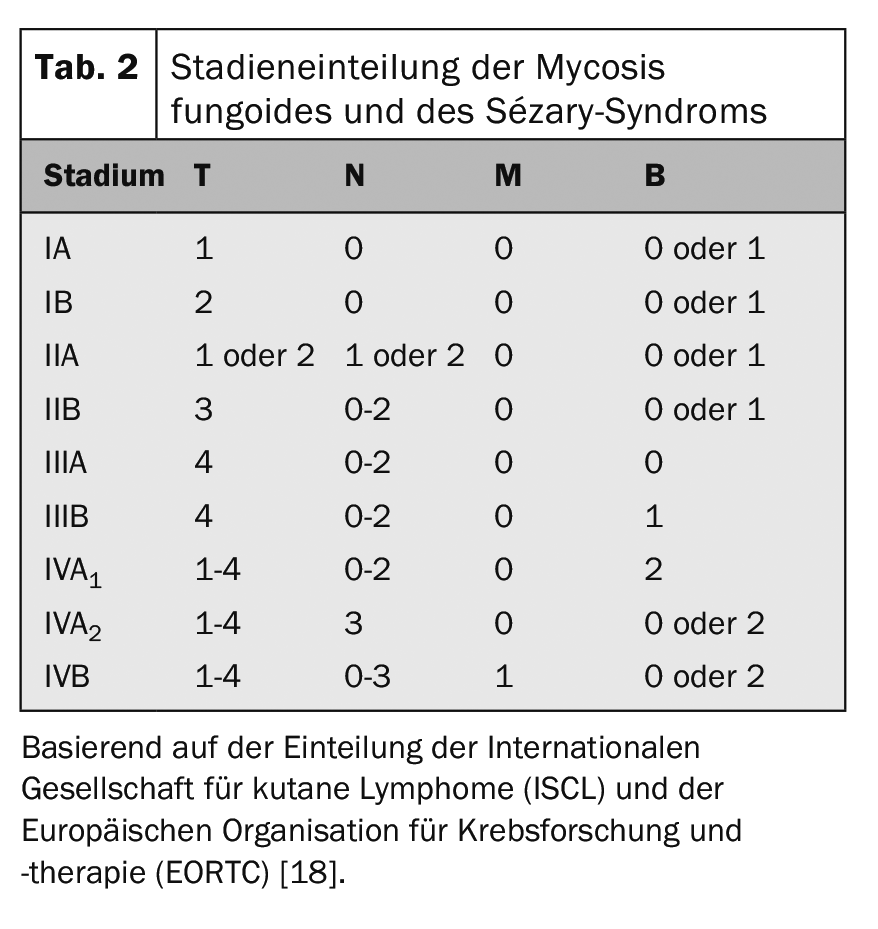
Per la stadiazione del tumore del CTCL si utilizza la classificazione TNM, riconosciuta a livello internazionale (Tab. 1). Oltre all’interessamento cutaneo – tumore primario (T), coinvolgimento linfonodale (N) e metastasi a distanza, questo include (M) – anche il rilevamento di cellule di Sézary nel sangue periferico (B) e quindi si parla di classificazione TNMB (tab. 1). Questa classificazione viene utilizzata per classificare le fasi (Tab. 2). La sindrome di Sézary con il suo coinvolgimento ematico obbligatorio corrisponde quindi sempre allo stadio IV [16]. Di conseguenza, la malattia ha una prognosi sfavorevole, con un tasso di sopravvivenza mediano di <3 anni. Oltre al coinvolgimento del sangue periferico, sono stati identificati altri fattori prognostici negativi: fenotipi atipici e la caratteristica perdita delle caratteristiche di superficie CD7 e CD26 [17].
Gli esami di diagnostica per immagini, come la TAC dell’intero corpo e la sonografia linfonodale come parte della stadiazione iniziale del tumore, sono raccomandati per tutti i pazienti dallo stadio T2 in poi [8,20]. La PET-CT e la biopsia linfonodale devono essere eseguite nei pazienti con sospetta linfoadenopatia e/o diffusione sistemica [8,16]. I pazienti con anomalie ematologiche devono sottoporsi a una biopsia del midollo osseo. La diagnostica avanzata e le biopsie degli organi viscerali possono essere prese in considerazione se si sospetta un coinvolgimento extracutaneo [8]. I pazienti affetti da sindrome di Sézary dovrebbero idealmente essere gestiti sia dal punto di vista diagnostico che terapeutico da un team multidisciplinare composto da dermatologi, oncologi, dermatopatologi e radiologi [16].
Eziologia
Sebbene la causa di questa complessa malattia non sia ancora del tutto chiarita, si può ipotizzare che la fisiopatologia della sindrome di Sézary possa essere spiegata da un lato da una disfunzione immunitaria e dall’altro da cambiamenti epigenetici [21]. Fino a poco tempo fa, si pensava che il cariotipo della sindrome di Sézary fosse caratterizzato da delezioni dei cromosomi 10q e 17p e da inserzioni dei cromosomi 8q, 10p e 17q [8]. Uno studio di Izykowska et al. mostra che le alterazioni maligne, tuttavia, si verificano ad un’ampia varietà di livelli genetici [21,22]. Come per la maggior parte dei CTCL leucemici, nella sindrome di Sézary c’è una risposta immunitaria prevalentemente Th2-pesante [23]. Le citochine Th2-tipiche interleuchina 4 (IL-4), IL-5, IL-13, IL-21 e IL-31, che vengono rilasciate in quantità elevate, sopprimono la risposta immunitaria mediata dai Th1 e fungono anche da bersaglio farmacologico [3,21].
Diagnosi differenziale
A livello differenziale, la sindrome di Sézary è particolarmente difficile da distinguere dalla micosi fungoide (MF), la più comune di tutte le CTCL. Dal punto di vista clinico e diagnostico, entrambe le malattie presentano molte somiglianze. Campbell et al. Tuttavia, sono riusciti a dimostrare che la sindrome di Sézary e la MF derivano da sottoinsiemi funzionali di cellule T diversi e che quindi dovrebbero essere considerati linfomi separati [3,24]. Mentre il coinvolgimento del sangue è assente o minimo nella MF, è una componente essenziale nella sindrome di Sézary [7]. Altre diagnosi differenziali non neoplastiche della sindrome di Sézary includono l’eritrodermia psoriatica, la dermatite atopica o altre forme di dermatite, la pitiriasi rubra pilaris, le reazioni ai farmaci e l’eritroderma idiopatico. Distinguere tra la sindrome di Sézary in fase iniziale e le dermatosi infiammatorie eritrodermiche può essere impegnativo.
Terapia
Il trattamento della sindrome di Sézary, che di solito non porta ancora a una guarigione completa, si basa principalmente sull’estensione della malattia, sull’influenza sulla qualità della vita, sui fattori prognostici e sull’età del paziente o sulle sue comorbidità [16]. Secondo l’ultimo aggiornamento terapeutico sulla sindrome di Sézary del JDDG, la fotoferesi extracorporea (ECP), che ha pochi effetti collaterali, è una delle misure terapeutiche di prima scelta. L’ECP può essere utilizzato come monoterapia o in combinazione con corticosteroidi topici, fototerapia con psoraleni e UV-A (PUVA), o per via sistemica con interferone alfa (INF-α) o bexarotene [19,26].
Come terapia di “seconda linea” si raccomandano basse dosi di metotrexato, retinoidi somministrati per via sistemica (bexarotene; entrambi anche in combinazione con PUVA ed ECP), il clorambucile, attivo dal punto di vista citostatico, in combinazione con un glucocorticoide a basso dosaggio, e l’irradiazione elettronica della pelle intera [19,20,26]. Negli stadi avanzati della sindrome di Sézary, la monochemioterapia con gemcitabina o doxorubicina liposomiale pegilata è una delle terapie di seconda linea.
Negli ultimi anni, il successo terapeutico è migliorato sempre di più grazie alla terapia oncologica mirata. Alemtuzumab, un anticorpo monoclonale umanizzato anti-CD52, è stato studiato in uno studio di fase II su 22 pazienti con MF/SS avanzata, con tassi di risposta del 55% [27].
A causa del rischio significativamente maggiore di complicazioni infettive, la somministrazione sottocutanea del farmaco a bassa dose è stata studiata in una coorte di 14 pazienti con SS refrattaria , con tassi di risposta superiori all’80% [28]. Il coniugato anticorpo-farmaco brentuximab vedotin non è ancora approvato per la sindrome di Sézary, ma può essere utilizzato come preparazione off-label per le varianti CD30+ della sindrome di Sézary. L’antagonista dell’acido folico pralatrexate è approvato per il linfoma a cellule T periferiche. Uno studio clinico randomizzato di fase III che ha confrontato l’anticorpo monoclonale CCR4 mogamulizumab con vorinostat (MAVORIC) ha mostrato un miglioramento significativo della sopravvivenza libera da progressione nei pazienti con MF / SS randomizzati a mogamulizumab. I pazienti con sindrome di Sézary hanno avuto la risposta complessiva più alta (37%) [29].
Diversi inibitori dell’istone deacetilasi (HDAC) che interferiscono con la regolazione epigenetica della trascrizione sono già stati approvati fuori dall’Europa per il trattamento della sindrome di Sézary [20]. In Europa, l’inibitore HDAC resminostat è attualmente in fase di sperimentazione clinica come terapia di mantenimento per i pazienti con SS avanzata e MF rispetto al placebo. Gli inibitori del checkpoint immunitario anti-PD1 e anti-CTLA4 sono ora approvati per il trattamento di molteplici tumori solidi, compreso il melanoma. I dati preliminari di uno studio di fase II di pembrolizumab (anticorpo monoclonale anti-PD1) nella MF recidivata/refrattaria e nella SS appaiono promettenti con un tasso di risposta del 33%, anche nei pazienti con malattia avanzata [30].
Attualmente, l’unica opzione di trattamento con potenziale curativo è il trapianto di cellule staminali allogeniche. Sebbene con questa terapia si possano osservare remissioni a lungo termine, l’aumento della mortalità e della morbilità associate al trapianto non deve essere sottovalutato [16,20]. Nonostante tutti i progressi scientifici, il trattamento della sindrome di Sézary, a parte il trapianto di cellule staminali, rimane tuttora palliativo.
Nonostante la prognosi attualmente infausta di questa malattia, c’è la speranza che le recenti scoperte relative alla sindrome di Sézary come quadro clinico indipendente e i progressi diagnostici e terapeutici associati migliorino le possibilità di guarigione e la qualità di vita delle persone colpite.
Messaggi da portare a casa
- Solo il 25,5% dei pazienti con sindrome di Sézary presenta un eritroderma classico come manifestazione iniziale.
- Il coinvolgimento del sangue è una componente essenziale della sindrome di Sézary e la distingue da altri linfomi cutanei primari.
- I sintomi non specifici della malattia nelle prime fasi portano a una significativa latenza diagnostica.
- La fotoferesi extracorporea e le nuove terapie mirate (ad esempio, utilizzando anticorpi monoclonali umanizzati) stanno migliorando sempre più il successo terapeutico nella sindrome di Sézary.
- Il trapianto di cellule staminali allogeniche è attualmente l’unica opzione di trattamento con potenziale curativo.
Letteratura:
- Swerdlow SH, Campo E, et al: La revisione 2016 della classificazione delle neoplasie linfoidi dell’Organizzazione Mondiale della Sanità. Sangue 2016; 127(20): 2375-2390.
- Scarisbrick JJ, Prince HM, et al: Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J Clin Oncol 2015; 33(32): 3766-3773.
- Saulite I, Hoetzenecker W, et al: Sindrome di Sézary e Dermatite Atopica: confronto tra aspetti immunologici e bersagli. Biomed Res Int 2016; articolo id: 9717530.
- Bradford PT, SS D, et al: Modelli di incidenza del linfoma cutaneo negli Stati Uniti: uno studio basato sulla popolazione di 3884 casi. Sangue 2009; 113(21): 5064-5073.
- Wilson LD, Hinds G, Yu JB: Età, razza, sesso, stadio e incidenza del linfoma cutaneo. Clin Lymphoma Myeloma Leuk 2012; 12(5): 291-296.
- Vonderheid EC, Bernengo MG, et al: Aggiornamento sul linfoma cutaneo a cellule T eritrodermico: rapporto della Società Internazionale per i Linfomi Cutanei. J Am Acad Dermatol 2002; 46(1): 95-106.
- Mangold AR, Thompson AK et al: Manifestazioni cliniche precoci della sindrome di Sézary: uno studio di coorte retrospettivo multicentrico. J Am Acad Dermatol 2017; pii: S0190-9622(17): 31784-X. doi: 10.1016/j.jaad.2017.05.036. [Epub ahead of print].
- Foss FM, Girardi M: Micosi Fungoide e sindrome di Sézary. Hematol Oncol Clin North Am 2017; 31(2): 297-315.
- Stevens SR, Ke MS, et al: Quantificazione dell’onere della malattia cutanea nei linfomi cutanei a cellule T di tipo micosi fungoide: lo strumento di valutazione ponderata per la gravità (SWAT). Arch Dermatol 2002; 138(1): 42-48.
- Trotter MJ, Whittaker SJ, Orchard GE, Smith NP: Istopatologia cutanea della sindrome di Sézary: uno studio di 41 casi con un clone di cellule T circolanti dimostrato. J Cutan Pathol 1997; 24(5): 286-291.
- Boonk SE, Zoutman WH, et al: Valutazione dei biomarcatori immunofenotipici e molecolari della sindrome di Sézary mediante procedure operative standard: Uno studio multicentrico su 59 pazienti. J Invest Dermatol 2016; 136(7): 1364-1372.
- Gibson JF, Huang J et al: Linfoma cutaneo a cellule T (CTCL): pratiche attuali nella valutazione del sangue e utilità della restrizione della catena del recettore delle cellule T (TCR)-Vbeta. J Am Acad Dermatol 2016; 74(5): 870-877.
- Lukowsky A, Muche JM et al.: Valutazione della clonalità delle cellule T in campioni di biopsia cutanea d’archivio di linfomi cutanei a cellule T, utilizzando il protocollo biomed-2 PCR. Diagn Mol Pathol 2010; 19(2): 70-77.
- Guenova E, Ignatova D et al: Espressione del CD164 sulle cellule T maligne nella sindrome di Sézary. Acta Derm Venereol 2016; 96(4): 464-467.
- Benoit BM, Jariwala N, et al: Il CD164 identifica le cellule T CD4+ che esprimono altamente i geni associati alla malignità nella sindrome di Sézary: i geni firma di Sézary, FCRL3, Tox e miR-214. Arch Dermatol Res 2017; 309(1): 11-19.
- Jawed SI, Myskowski PL, et al: Linfoma primario cutaneo a cellule T (micosi fungoide e sindrome di Sézary): parte II. Prognosi, gestione e indicazioni future. J Am Acad Dermatol 2014; 70(2): 223.e1-17.
- Scarisbrick JJ, Kim YH, et al: Fattori prognostici, indici prognostici e stadiazione nella micosi fungoide e nella sindrome di Sézary: a che punto siamo? Br J Dermatol 2014; 170(6): 1226-1236.
- Olsen E, Vonderheid E, Pimpinelli N, et al: Revisioni della stadiazione e della classificazione della micosi fungoide e della sindrome di Sézary: una proposta della Società Internazionale per i Linfomi Cutanei (ISCL) e della task force sui linfomi cutanei dell’Organizzazione Europea per la Ricerca e il Trattamento del Cancro (EORTC). Sangue 2007; 110(6): 1713-1722.
- Dippel E, Assaf C, Becker J, Bergwelt M, Beyer M: S2k – Linea guida breve – Linfomi cutanei (ICD10 C82 – C86) Aggiornamento 2016. JDDG 2017; [in Vorbereitung].
- Trautinger F, Eder J, et al: Raccomandazioni di consenso dell’Organizzazione Europea per la Ricerca e la Cura del Cancro per il trattamento della micosi fungoide/sindrome di Sézary – Aggiornamento 2017. Eur J Cancer 2017; 77: 57-74.
- DeSimone JA, Sodha P, Ignatova D, et al: Recenti progressi nel linfoma primario a cellule T cutaneo. Curr Opin Oncol 2015; 27(2): 128-133.
- Izykowska K, Przybylski GK, et al.: I riarrangiamenti genetici determinano un’alterazione dell’espressione genica e nuovi trascritti di fusione nella sindrome di Sézary. Oncotarget 2017; 8(24): 39627-39639.
- Guenova E, Watanabe R et al: Le citochine TH2 delle cellule maligne sopprimono le risposte TH1 e rafforzano una polarizzazione TH2 globale nel linfoma cutaneo a cellule T leucemico. Clin Cancer Res 2013; 19(14): 3755-3763.
- Campbell JJ, Clark RA, et al: La sindrome di Sézary e la micosi fungoide derivano da sottogruppi di cellule T distinti: un fondamento biologico per i loro comportamenti clinici distinti. Sangue 2010; 116(5): 767-771.
- Willemze et al: L’aggiornamento 2018 della classificazione WHO-EORTC per i linfomi cutanei primari, Blood (2019) 133 (16): 1703-1714.
- Dippel E, et al: S2k-Leitlinie – Kutane Lymphome Update 2016 – Teil 2: Therapie und Nachsorge (ICD10 C82 – C86), J Dtsch Dermatol Ges. 2018 Jan;16(1): 112-123.
- Lundin J, Hagberg H, Repp R, et al. Studio di fase 2 di alemtuzumab (anticorpo monoclonale anti-CD52) nei pazienti con micosi fungoide/sindrome di Sezary in fase avanzata. Sangue. 2003;101: 4267-4272.
- Bernengo MG, Quaglino P, Comessatti A, et al: Alemtuzumab intermittente a basso dosaggio nel trattamento della sindrome di Sezary: risultati clinici e immunologici in 14 pazienti. Haematologica 2007; 92: 784-794.
- Kim YH, et al: Mogamulizumab vs vorinostat nel linfoma cutaneo a cellule T precedentemente trattato (MAVORIC): uno studio internazionale di fase 3, in aperto, randomizzato e controllato. Lancet Oncol. 2018;19: 1192-1204).
- Phillips T, Devata S, Wilcox RA: Sfide e opportunità per il blocco del checkpoint nei disordini linfoproliferativi a cellule T. J Immunother Cancer 2016; 4: 95.
PRATICA DERMATOLOGICA 2019; 29(6): 14-18