Tipico dell’amiloidosi cardiaca è lo sviluppo di insufficienza cardiaca con frazione di eiezione preservata (HFpEF). Nuove procedure diagnostiche e opzioni terapeutiche stanno ora gettando una luce diversa su questa malattia rara, finora praticamente incurabile.
Con l’introduzione di nuove procedure diagnostiche e opzioni terapeutiche per quella che è stata una malattia rara praticamente incurabile, l’interesse per l’amiloidosi sta crescendo rapidamente. Il coinvolgimento cardiaco (amiloidosi cardiaca) di solito porta a una grave insufficienza cardiaca con tutte le sue conseguenze cliniche. L’obiettivo di questo articolo è di fornire una breve panoramica della moderna diagnosi e terapia dell’amiloidosi cardiaca.
Origine
Nell’amiloidosi sistemica, ci sono depositi di proteine ripiegate in modo anomalo e aggregate nel tessuto. A seconda dell’infestazione dell’organo, si presentano i disturbi funzionali e le malattie corrispondenti. Se i depositi si trovano nel cuore (nell’interstizio tra le cellule del miocardio), questo porta a una malattia cardiaca con il quadro clinico dell’insufficienza cardiaca. Nel cuore si depositano solitamente due tipi diversi di amiloide, a seconda della proteina antecedente difettosa.
Amiloidosi AL: di solito si sviluppa sulla base di una discrasia delle plasmacellule. Le cellule plasmatiche clonali (ad esempio nel mieloma multiplo) producono un eccesso di catene leggere libere. Nel caso delle catene leggere amiloidogeniche, queste si depositano come fibrille amiloidi in vari organi. Il coinvolgimento cardiaco isolato è raro nell’amiloidosi AL. Molto spesso vengono colpiti anche i reni, il fegato e il sistema nervoso periferico. Le catene leggere Lambda sono rilevate molto più frequentemente come componenti amiloidi rispetto alle catene kappa.
Amiloidosi ATTR: la transtiretina (TTR) è una proteina di trasporto, prodotta per il 98% nel fegato e responsabile del trasporto della tiroxina e del retinolo. La TTR è un tetramero che può dissociarsi in quattro monomeri. Normalmente, questi monomeri sono solubili nel sangue. Tuttavia, se c’è un disturbo, i monomeri si aggregano per formare fibrille amiloidi patologiche. Esistono due cause di amiloidosi TTR: la forma mutata (ereditaria, familiare) si verifica a causa di una mutazione nel gene TTR. La prevalenza delle mutazioni varia a seconda della regione geografica e dei gruppi etnici. La forma “wild-type” (acquisita, senile) di solito si manifesta solo in età avanzata e colpisce prevalentemente il cuore.
Manifestazione clinica
Coinvolgimento cardiaco: tipico dell’amiloidosi cardiaca è lo sviluppo di insufficienza cardiaca con frazione di eiezione preservata (HFpEF). I depositi di amiloide rendono le pareti dei ventricoli e degli atri più spesse (non c’è un’ipertrofia vera e propria, poiché le cellule del muscolo cardiaco di per sé non sono interessate). Questo porta a un “irrigidimento” del cuore e a una restrizione del rilassamento (disfunzione diastolica). Pertanto, si verifica un aumento delle pressioni di riempimento end-diastoliche nel ventricolo, una dilatazione degli atri e un aumento della pressione polmonare. Clinicamente, questo porta alla fine a un’insufficienza cardiaca destra e sinistra con dispnea, intolleranza alle prestazioni ed edema. È anche tipico che il ventricolo rigido non riesca più a regolare correttamente la gittata cardiaca. Poiché il volume della corsa è fissato dalla rigidità, la gittata cardiaca può essere controllata praticamente solo dalla frequenza cardiaca. Possono quindi verificarsi sincope e grave disregolazione ortostatica, soprattutto sotto stress, in particolare nei pazienti che assumono beta-bloccanti.
L’atrio sinistro allargato è un substrato “buono” per lo sviluppo della fibrillazione atriale. Le pressioni elevate, insieme alla bassa mobilità dell’atrio a causa dei depositi di amiloide, possono portare ad un aumento del rischio di trombi intra-atriali e spiegare il rischio significativamente aumentato di infarti cerebrali nei pazienti con amiloidosi.
Se i depositi di amiloide colpiscono il sistema di conduzione, si verificano dei blocchi, soprattutto quelli AV. Inoltre, aumenta il rischio di morte cardiaca improvvisa dovuta ad aritmie ventricolari (soprattutto fibrillazione ventricolare e “attività elettrica senza polso” – PEA).
Un’altra manifestazione tipica dell’amiloidosi cardiaca è il dolore toracico, senza che venga riscontrata una vera e propria malattia coronarica. Da un lato, piccole microtrombosi nel microcircolo possono scatenare l’angina pectoris (spasmi); dall’altro, la funzione endoteliale disturbata rivela una disfunzione microvascolare.
Coinvolgimento di altri organi: il frequente problema di ortostasi nei pazienti affetti da amiloidosi è solitamente causato da un danno al sistema nervoso periferico e dall’associata ridotta vasocostrizione dei vasi periferici.
Un’innervazione intestinale disturbata può portare a disturbi del transito, flatulenza, dolore addominale e irregolarità delle feci. Il coinvolgimento dei reni si manifesta di solito con proteinuria e diminuzione della funzionalità renale.
I depositi di amiloide nel tratto gastrointestinale causano occasionalmente malassorbimento, perdita di peso e aumento della rigidità del fegato. Nell’amiloidosi AL, occasionalmente (10%) si possono riscontrare i segni patognomonici di emorragia periorbitale e macroglossia. L’amiloidosi TTR è spesso preceduta dalla sindrome del tunnel carpale.
Diagnostica
Se si sospetta clinicamente l’amiloidosi cardiaca, si utilizzano diverse procedure diagnostiche. L’ECG spesso mostra un basso voltaggio periferico (attenuazione della conduzione elettrica verso gli elettrodi a causa della deposizione di amiloide), una caratteristica che lo distingue dalla cardiomiopatia ipertrofica, dove di solito si osserva un aumento del voltaggio. Si possono vedere anche immagini di blocco, fibrillazione atriale e altre aritmie. Tuttavia, tutti questi cambiamenti non sono specifici della malattia. Oltre ai parametri abituali, è necessario misurare in laboratorio i biomarcatori cardiovascolari NT-proBNP e troponina. Non deve mancare un esame delle urine, soprattutto per quanto riguarda l’albumina e l’urina. Anche l’elettroforesi delle proteine e l’immunofissazione nel siero e nelle urine sono essenziali per distinguere le forme AL e ATTR.
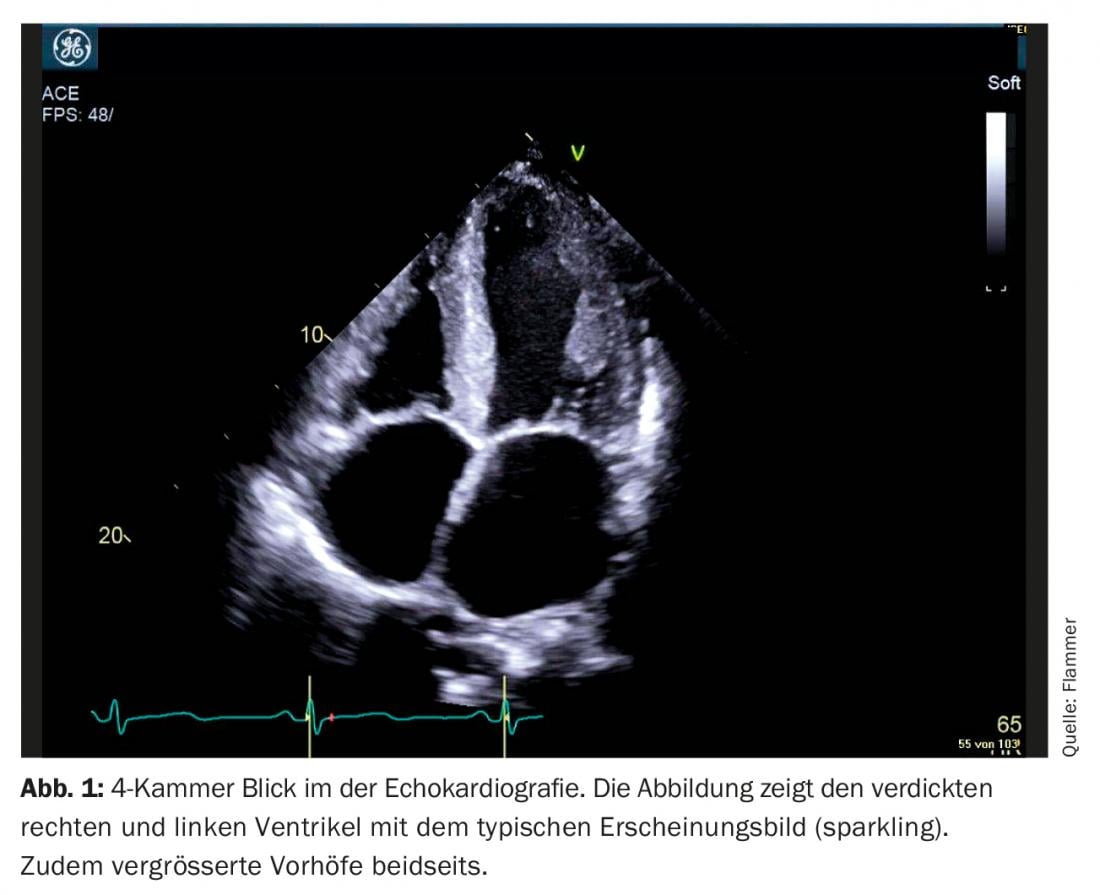
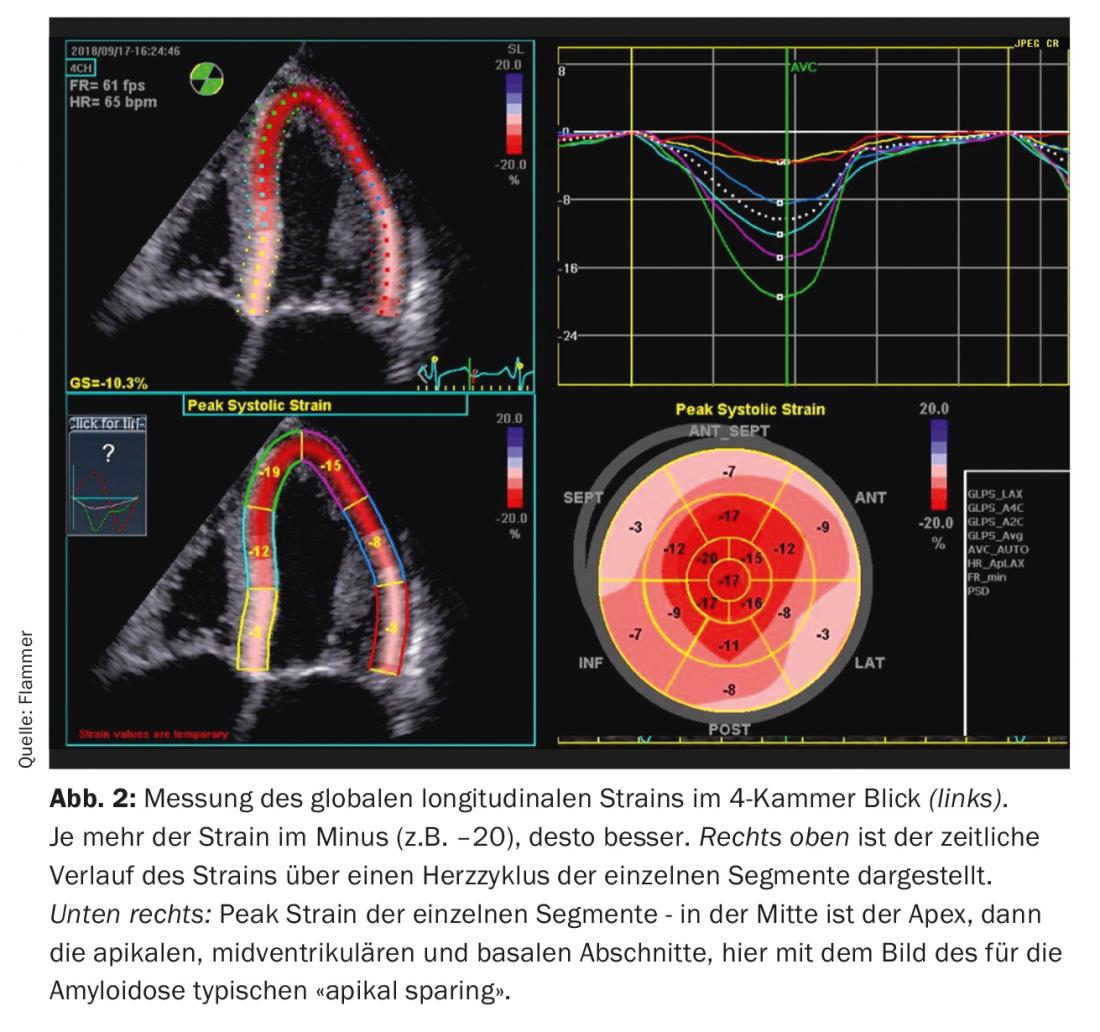
Ecocardiografia: la diagnosi di amiloidosi cardiaca è impensabile senza l’imaging cardiaco. I pazienti con un’anamnesi e segni di insufficienza cardiaca di solito vengono sottoposti a ecocardiografia. Se ci sono segni corrispondenti di amiloidosi nell’ecografia e un’insufficienza cardiaca manifesta, l’amiloidosi deve essere presa in considerazione. Quali sono i segni tipici dell’ecocardiografia? C’è un miocardio biventricolare ispessito, e anche il setto intraatriale e le pareti atriali sono solitamente ispessite. Il miocardio si presenta anche in modo specifico come “scintillante”, che è caratteristico della malattia (tuttavia, è importante ricordare che la “seconda immagine armonica” deve essere disattivata sull’ecografo). L’amiloide si deposita anche sulle valvole. Pertanto, appaiono leggermente ispessite, ma la funzione delle valvole è solitamente normale. Anche un discreto versamento pericardico è comune (Fig. 1). Dal punto di vista funzionale, soprattutto nella malattia avanzata, c’è una grave disfunzione diastolica, di solito con un modello di riempimento restrittivo. A causa dell’aumento delle pressioni diastoliche associate, entrambi gli atri si allargano nel corso della malattia (dilatazione biatriale) – caratteristica della disfunzione diastolica grave. Sebbene la frazione di eiezione sia solitamente normale, c’è anche una disfunzione sistolica, che si manifesta con una “deformazione” ridotta (la deformazione descrive la deformazione misurata del miocardio). In particolare, la “deformazione” longitudinale globale è limitata. Tipico e specifico per l’amiloidosi è il cosiddetto “sparing apicale”, cioè il ceppo è normale (risparmiato) nell’apice, ma si deteriora più si va verso la base del cuore (Fig. 2).
Risonanza magnetica (RM): al giorno d’oggi, l’amiloidosi viene spesso diagnosticata con questo esame (a volte come risultato incidentale in altre indagini). I cambiamenti osservati nella risonanza magnetica sono simili in linea di principio a quelli osservati nell’ecografia, ma si possono notare ulteriori cambiamenti tipici, soprattutto se si utilizza anche il mezzo di contrasto gadolinio. A causa di un lento wash-out del gadolinio, questo rimane più a lungo nell’interstizio se è presente l’amiloide. La risonanza magnetica mostra poi un “potenziamento tardivo del gadolinio” diffuso e a chiazze, tipico dell’amiloidosi.
Algoritmo diagnostico per il sospetto di amiloidosi cardiaca
Nei pazienti con un quadro clinico corrispondente e un sospetto di amiloidosi cardiaca, si deve innanzitutto escludere la discrasia plasmacellulare. Per questo, è necessario richiedere l’elettroforesi delle proteine e l’immunofissazione nelle urine e nel siero, come descritto sopra. Se è presente, la ricerca è principalmente per l’amiloidosi AL e, in assenza di segni, per l’amiloidosi ATTR (Fig. 3).
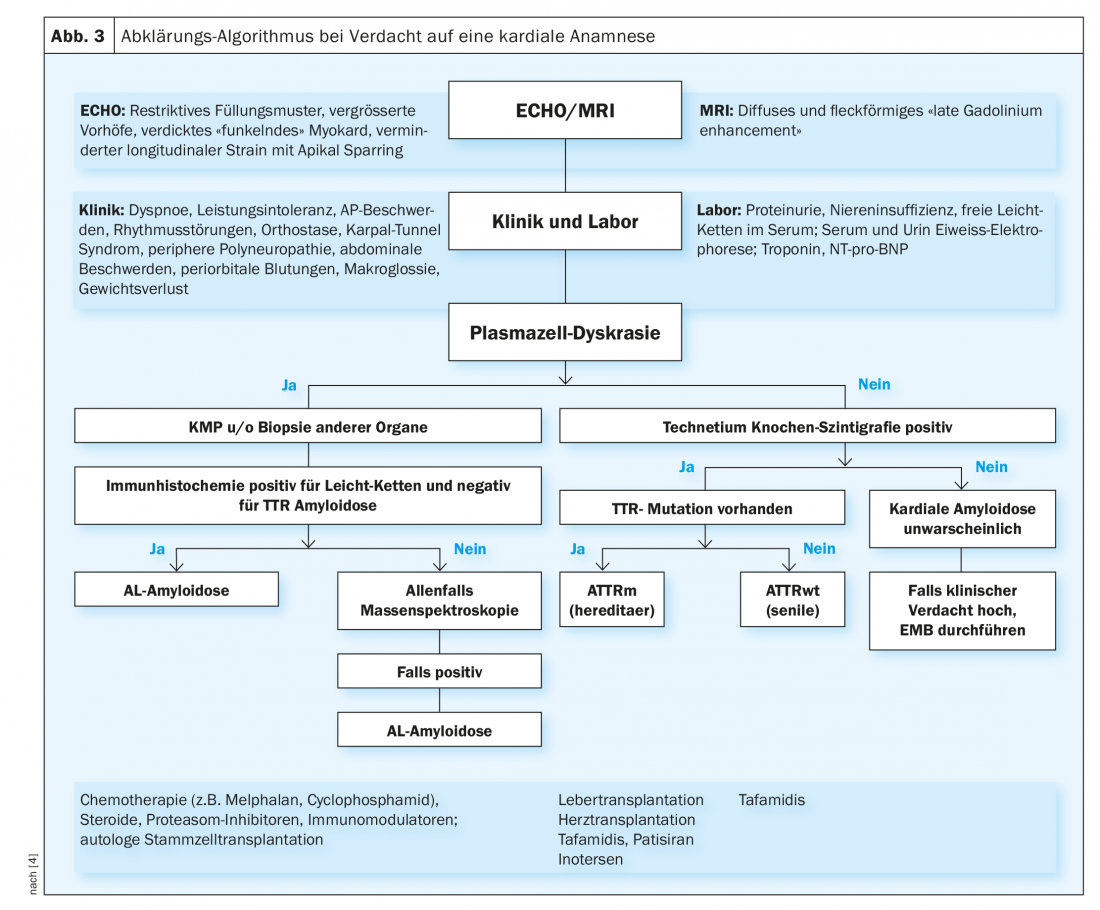
Se si sospetta l’amiloidosi AL (è presente una discrasia delle plasmacellule), si deve cercare di rilevare l’amiloide AL direttamente per via biotopica. Un’aspirazione del midollo osseo viene solitamente eseguita per diagnosticare la malattia ematologica di base (di solito il mieloma multiplo). Poiché l’amiloidosi è una malattia sistemica, la diagnosi è confermata non appena viene rilevata l’amiloide AL in una biopsia (di qualsiasi tessuto). Il metodo meno invasivo e traumatico è l’aspirazione del tessuto adiposo addominale. Inoltre, a volte l’amiloide può essere ricercata in un prelievo di tessuto eseguito in precedenza, nell’ambito di una colonscopia o di una gastroscopia. Inoltre, possono essere eseguite biopsie delle ghiandole salivari, delle labbra o del retto. Occasionalmente, è necessaria anche una biopsia renale o endomiocardica per fare una diagnosi definitiva. Nell’amiloidosi cardiaca, la biopsia endomiocardica è molto sensibile. Da un lato, le biopsie vengono esaminate per verificare la presenza di amiloide e, dall’altro, viene utilizzata l’immunoistochimica per cercare di classificare il tipo di amiloidosi. Nei casi difficili, è necessaria la spettroscopia di massa (gold standard). Tuttavia, solo poche patologie prevedono un esame di questo tipo.
Se si sospetta un’amiloidosi TTR (senza evidenza di discrasia plasmacellulare), oggi si esegue una scintigrafia al tecnezio (ossea). In queste condizioni, la sensibilità e la specificità di questa metodologia nell’amiloidosi ATTR cardiaca sono molto buone. Se i risultati sono positivi, si può quindi omettere una biopsia e fare la diagnosi. Se la scintigrafia non è chiara e si sospetta ancora un’amiloidosi cardiaca, è necessario eseguire una biopsia endomiocardica in questo sito.
Una volta confermata l’amiloidosi ATTR, è necessario un test genetico per distinguere se si tratta di una forma “wild-type” o ereditaria.
Terapia dell’amiloidosi cardiaca
L’insufficienza cardiaca è di solito al centro dei sintomi dell’amiloidosi cardiaca. Dal punto di vista terapeutico, questi aspetti devono essere affrontati prima di tutto. I diuretici aiutano a livello sintomatico. Soprattutto perché le pressioni di riempimento cardiaco sono elevate a causa della fisiologia cardiaca restrittiva, che porta all’innalzamento della pressione venosa polmonare passiva. I pazienti sono significativamente meno sintomatici con i diuretici. Nel corso della malattia, di solito sono necessarie dosi sempre più elevate – di conseguenza, è anche importante assicurarsi di sostituire una quantità sufficiente di potassio. La titolazione della dose è spesso impegnativa (riquadro).

Secondo la fisiologia dell’HFpEF, non vi è alcuna indicazione per gli ACE-inibitori, i beta-bloccanti e gli antagonisti dell’aldosterone. Al contrario, gli ACE-inibitori in particolare possono esacerbare quella che spesso è un’ortostasi già pronunciata. I beta-bloccanti comportano l’incapacità di aumentare la gittata cardiaca sotto sforzo, a causa del volume di ictus fisso. Pertanto, i beta-bloccanti vengono sospesi per i pazienti con fibrillazione atriale tachicardica.
La fibrillazione atriale e gli infarti cerebrali che ne derivano sono comuni. Da un lato, è essenziale non perdere la VHF (esami Holter semestrali), dall’altro, l’anticoagulazione orale deve essere iniziata se presente, indipendentemente dal punteggio CHADS-Vasc. Non è del tutto chiaro se l’OAK debba essere iniziato in assenza di VCF – è probabile che questo sia appropriato nel caso di un modello di riempimento restrittivo. Nella VHF tachicardica, si deve controllare la frequenza con un beta-bloccante; la digossina non deve essere somministrata a causa dell’aumento della tossicità. L’amiodarone è indicato in alcuni pazienti per il controllo del ritmo e occasionalmente per il controllo della frequenza.
A causa dei disturbi del sistema di conduzione cardiaca, l’impianto di pacemaker diventa necessario, soprattutto nei pazienti con amiloidosi TTR, un’indicazione che può essere data generosamente. La questione dell’opportunità di impiantare un defibrillatore cardioverter impiantabile (ICD) è particolarmente impegnativa. La causa più comune di morte cardiaca nei pazienti con amiloidosi è la morte cardiaca improvvisa dovuta ad aritmie ventricolari. Non si osserva solo la fibrillazione ventricolare, ma anche un numero rilevante di PEA. Questi ultimi non vengono rilevati in modo affidabile da un ICD. C’è anche un tasso più elevato di shock applicati in modo errato (il che è molto traumatico per i pazienti) e di erogazioni di shock non riuscite. Finora esistono solo studi retrospettivi su questo argomento, che non mostrano alcun chiaro beneficio per un ICD. L’indicazione deve quindi essere fatta con cautela e discussa in dettaglio in un team di amiloidosi.
Terapia causale
Nell’amiloidosi AL, la malattia ematologica deve essere affrontata prima di tutto. Tuttavia, questo è spesso impegnativo, soprattutto nei casi di grave coinvolgimento cardiaco e di concomitante insufficienza cardiaca grave. La terapia ad alto dosaggio con trapianto di cellule staminali autologhe, a cui spesso si punta, non può essere effettuata in questo caso. Anche gli altri regimi terapeutici con agenti chemioterapici (ad esempio, melfalan, ciclofosfamide), steroidi e nuovi inibitori del proteasoma, immunomodulatori o anticorpi anti-CD38 possono occasionalmente essere stressanti per il cuore. Tuttavia, in alcuni casi esistono approcci curativi. Tuttavia, una trattazione dettagliata andrebbe oltre lo scopo di questo articolo.
Per l’amiloidosi TTR ereditaria, il trapianto di fegato (e il trapianto di cuore negli stadi avanzati) rimane il trattamento di scelta.
Tuttavia, dall’estate 2018, negli Stati Uniti e in Europa sono disponibili due farmaci (approvati dalla FDA e dall’EMA) che utilizzano la tecnologia di interferenza dell’RNA per ‘silenziare’ il gene difettoso, in modo che non venga prodotta la TTR amiloidogenica. Nessuno dei due farmaci è stato ancora approvato in Svizzera. Uno studio su larga scala condotto su pazienti affetti da amiloidosi ereditaria ha dimostrato in modo impressionante come la polineuropatia (spesso prevalente nell’amiloidosi ereditaria) possa essere fermata. Le analisi di sottogruppo del farmaco patisiran mostrano che i risultati nel cuore potrebbero essere altrettanto buoni, ma restano da vedere ulteriori studi. Fino a poco tempo fa, non esisteva una terapia causale per l’amiloidosi TTR di tipo selvaggio. Fortunatamente, uno studio pubblicato nell’estate del 2018 ha dimostrato che tafamidis, uno stabilizzatore della transtiretina, approvato anche in Europa e negli Stati Uniti per il trattamento della neurpatia amiloide ereditaria, ha buoni risultati nell’amiloidosi cardiaca, con un miglioramento della qualità di vita, una riduzione della mortalità e una riduzione delle ospedalizzazioni correlate al cuore. Attualmente il farmaco non è ancora disponibile in Svizzera, ma l’approvazione è prevista a breve, visti i buoni risultati.
Messaggi da portare a casa
- L’amiloidosi AL è una malattia rara che di solito è scatenata da una malattia ematologica sottostante che produce “catene leggere” e spesso colpisce altri organi oltre al cuore. Al centro della terapia c’è il trattamento della malattia ematologica.
- L’amiloidosi TTR può essere causata da una mutazione nel gene TTR o può essere “acquisita” nel corso della vita (“wild-type”). La forma familiare, più rara, si manifesta prevalentemente con polineuropatia o cardiopatia (o una combinazione), a seconda del gene coinvolto; la forma “wild-type” è solitamente limitata al cuore. Oltre alla terapia sintomatica, esistono nuovi approcci terapeutici promettenti con stabilizzatori della TTR e molecole di interferenza dell’RNA.
- La diagnosi di amiloidosi cardiaca richiede una clinica appropriata (insufficienza cardiaca), risultati ecografici o di risonanza magnetica tipici dell’amiloidosi e prove di amiloide in una biopsia o una scintigrafia Tc positiva.
- Il quadro clinico tipico dell’amiloidosi cardiaca comprende, in particolare, sintomi di insufficienza cardiaca, ortostatismo, sincope, aritmie (soprattutto VHF e tachicardia ventricolare/fibrillazione ventricolare), blocco AV e angina microvascolare.
Letteratura:
- Brouwers S, et al: Amiloidosi cardiaca Cardiovasc Med. 2018; 21(11): 282-289.
- Rauch PJ, et al: Amiloidosi sistemiche Switzerland Med Forum 2014 14; 943-948.
- Laptseva N, et al: Amiloidosi cardiaca: ancora una sfida Eur Heart J 2017, 38(22): 122.
- Falk RH, et al: Amiloidosi cardiaca AL (a catena leggera): una revisione della diagnosi e della terapia.
- Gillmore JD, et al: Diagnosi non bioptica di amiloidosi transtiretinica cardiaca Circulation 2016: j133(24): 2404-2412.
- Maurer MS, et al: Trattamento Tafamidis per i pazienti con cardiomiopatia amiloide transtiretina N Engl J Med 2018; 379(11): 1007-1016.
- Adams D, et al: Patisiran, un RNAi terapeutico, per l’amiloidosi transtiretinica ereditaria N Engl J Med 2018; 379(1): 11-21.
CARDIOVASC 2019; 18(2): 6-10