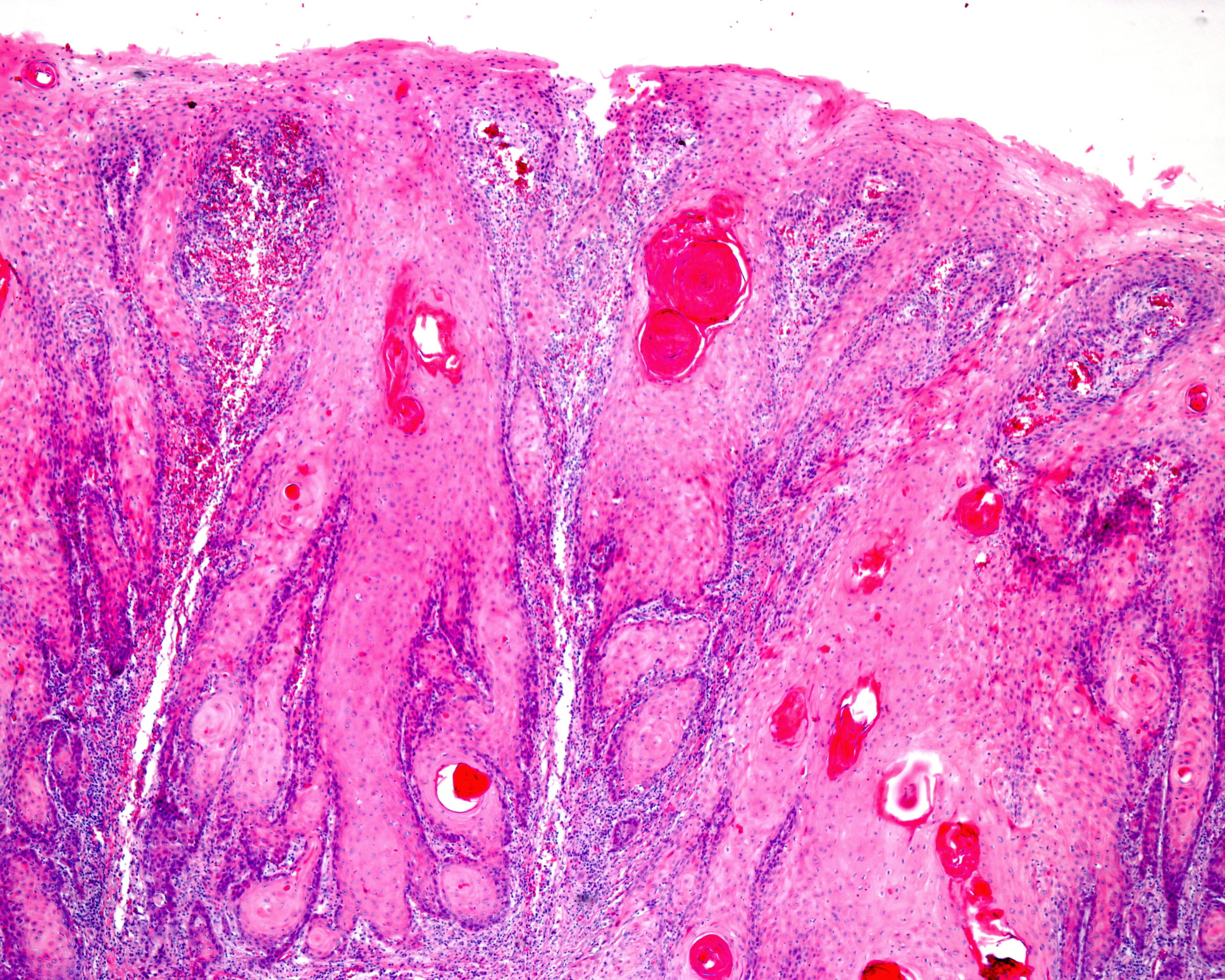
Il focus della terapia è ancora sulle opzioni di trattamento sintomatico e sull’evitare i fattori di provocazione. Le opzioni terapeutiche molecolari, potenzialmente anche curative, per l’epidermolisi bollosa sono ancora in fase di ricerca. Nel work-up diagnostico di questa malattia autoimmune eterogenea dal punto di vista genetico e fenotipico, è necessario considerare una serie di aspetti.
L’epidermolisi bollosa (EB) è un gruppo di genodermatosi eterogenee dal punto di vista genetico e fenotipico, caratterizzate da un’eccessiva fragilità meccanica dei tessuti epitelizzati. Con una prevalenza di circa 500.000 casi in tutto il mondo, l’EB è una malattia rara [1,2]. Ad oggi, sono state descritte mutazioni in oltre 20 geni che codificano componenti coinvolti nell’assemblaggio dei filamenti di cheratina del citoscheletro, molecole di adesione, desmosomi, emidesmosomi e fibrille di ancoraggio degli epiteli. (Fig. 1). Di conseguenza, l’integrità strutturale e funzionale dell’adesione intraepidermica e dell’adesione dermoepidermica alla pelle e alle membrane mucose viene compromessa, influenzando la funzione barriera, l’interazione cellula-cellula e cellula-matrice, la proliferazione, la rigenerazione dei tessuti e i processi di differenziazione. [3–5]. Lo spettro combinatorio del tipo, dell’omo- o dell’eterozigosi, del numero (eredità mono- o digenetica) e della localizzazione della mutazione nel rispettivo segmento genico, così come il conseguente disturbo quantitativo (assenza o riduzione) e qualitativo (perdita graduale della funzione) dell’espressione proteica, provoca considerevoli varianti geno- e di conseguenza fenotipiche dell’EB. Oltre al difetto genetico primario, anche i fattori epigenetici e biochimici secondari (ad esempio l’induzione di cascate infiammatorie croniche e il rimodellamento dei tessuti), così come i fattori ambientali, influenzano la gravità clinica [6,7].
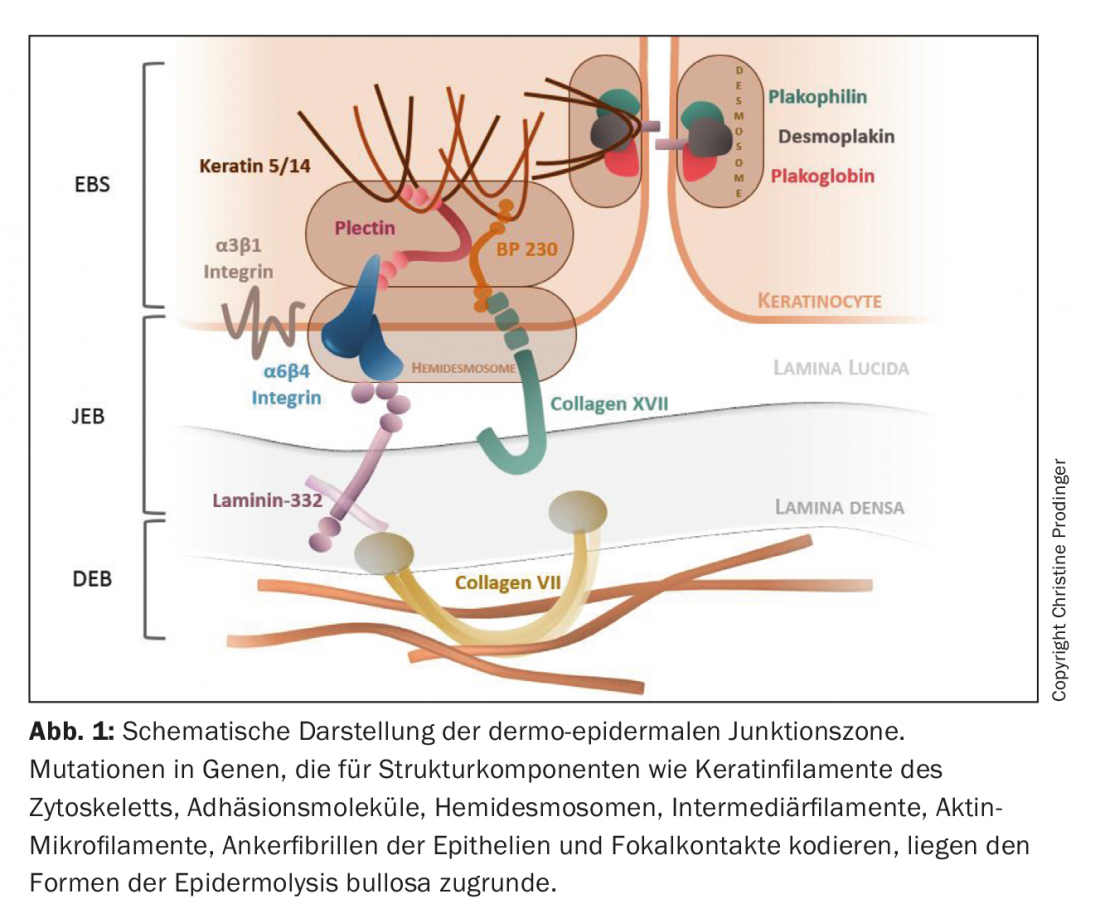
Poiché i geni indice interessati non sono espressi solo nella pelle e nelle membrane mucose vicine alla pelle, ma anche in altri epiteli (tratto respiratorio, urogenitale e gastrointestinale) e nel tessuto mesenchimale (muscoli scheletrici), sono possibili anche manifestazioni primarie extracutanee. L’EB può quindi trasformarsi in una malattia sistemica con morbilità e mortalità significative [8].
Classificazione fisiopatologica
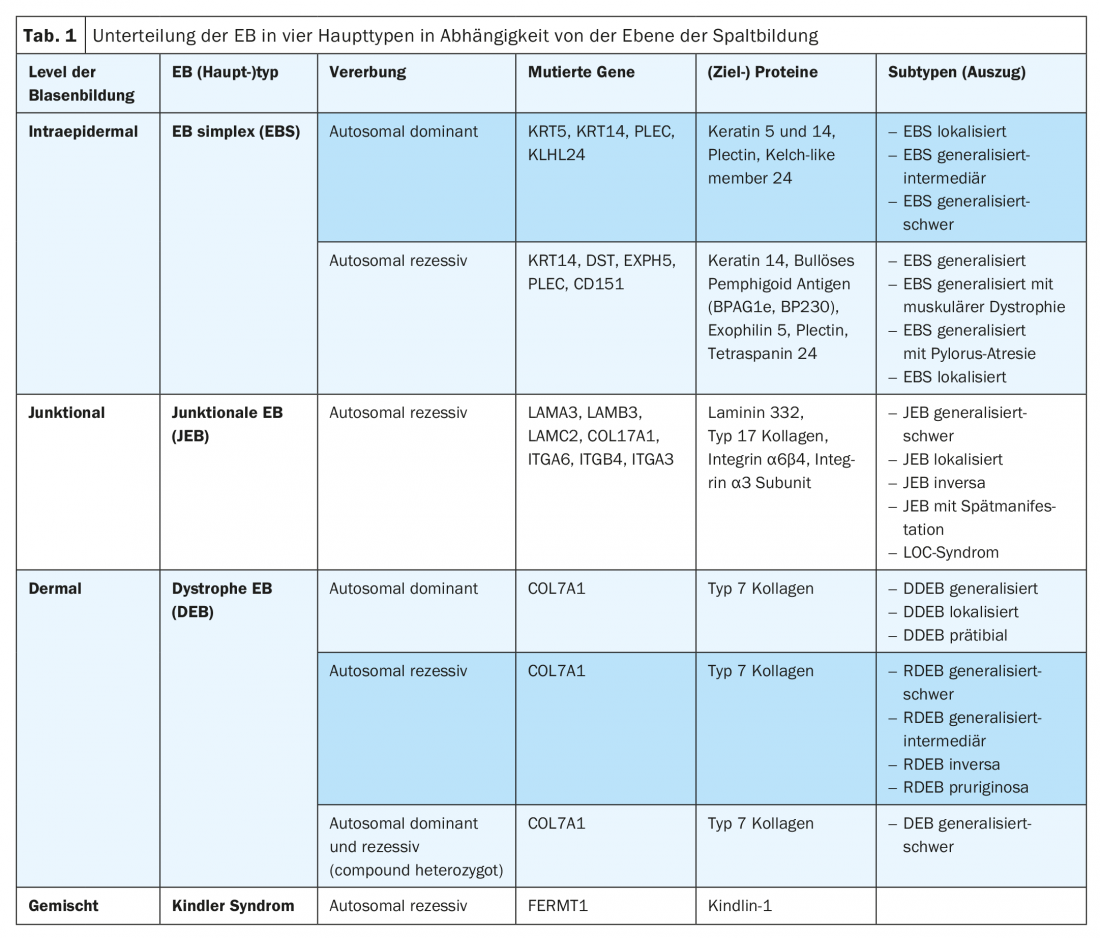
Il sintomo principale dell’EB è l’aumentata o eccessiva fragilità della pelle e delle membrane mucose nei confronti delle sollecitazioni meccaniche, con la formazione di vesciche, erosioni, ulcerazioni, croste o cicatrici, a seconda del sottotipo. In base al livello di scissione come funzione e conseguenza della mutazione sottostante, l’EB è suddivisa in quattro tipi principali (Tab. 1) [9]. Grazie alla crescente disponibilità di moderni metodi diagnostici molecolari (ad esempio, il “sequenziamento di nuova generazione”), vengono descritti sempre nuovi (sotto)tipi di EB. Quindi, una mutazione nel gene KLHL24 che codifica un componente del complesso della ligasi dell’ubiquitina potrebbe essere assegnata a una variante autosomica dominante dell’EB simplex, che comporta un’eccessiva ubiquitinazione e degradazione della cheratina 14 [10–12]. In un fenotipo simile alla sindrome di Kindler, è stata recentemente identificata anche una mutazione nel gene CD151, che codifica per una tetraspanina nella zona della membrana basale. Questa proteina transmembrana interagisce con le integrine ed è coinvolta nei processi di crescita, sviluppo e motilità cellulare [13]. Inoltre, di recente è stata scoperta una mutazione nel gene PLOD3, che codifica la lisil-idrossilasi 3 e regola l’elaborazione post-traslazionale del collagene di tipo 7. Dal punto di vista clinico, i soggetti colpiti presentano difetti estesi del tessuto connettivo, contratture articolari, malformazioni scheletriche e ritardo nella crescita. Il livello di formazione di vescicole è simile a quello dell’EB distrofica recessiva nella sublamina densa [14].
Algoritmo diagnostico
Soprattutto nel neonato con formazione di vesciche, si deve innanzitutto escludere una genesi traumatica, metabolica, ematologica, infettivologica, medicinale e autoimmune [15]. Successivamente, viene applicato un algoritmo diagnostico:
- Anamnesi (familiare) e clinica
- Determinazione della contaminazione microbica (ad esempio, strisci, PCR, sierologia).
- Biopsia perilesionale per la valutazione istologica di possibili diagnosi differenziali
- Immunofluorescenza diretta paralesionale per determinare il livello di clivaggio e l’espressione semiquantitativa della proteina da una vescica indotta (ad esempio, rotazione di una gomma da cancellare fino allo sviluppo di un eritema) e fresca (non più vecchia di 12 ore) su pelle non esposta al sole.
- Microscopia elettronica a trasmissione per determinare il piano di clivaggio e i difetti morfologici
- Analisi delle mutazioni (dipendente dal caso, come sequenziamento del genoma intero, dell’esoma, del cluster, del pannello)
Il rispettivo (sotto)tipo di EB può essere determinato in base ai risultati raccolti. Il piano di clivaggio determinato dalla microscopia elettronica a trasmissione e dall’immunofluorescenza definisce il tipo principale di EB. Il fenotipo clinico viene definito indicando la gravità relativa (lieve, intermedia, grave) e il modello di distribuzione (localizzato, generalizzato) – se opportuno, vengono indicati anche i sintomi caratteristici come la pseudosindattilia. Oltre ai risultati specifici raccolti nella microscopia elettronica a trasmissione o nella mappatura di immunofluorescenza, vengono elencati la proteina e il gene specifici colpiti, il tipo di mutazione o la mutazione specifica [9]. I successi nella caratterizzazione molecolare della base patogenetica dei diversi tipi di EB consentono anche una consulenza genetica, una prognosi e una diagnosi prenatale più precise e sono il presupposto fondamentale per interventi terapeutici innovativi e mirati.
Principi della terapia
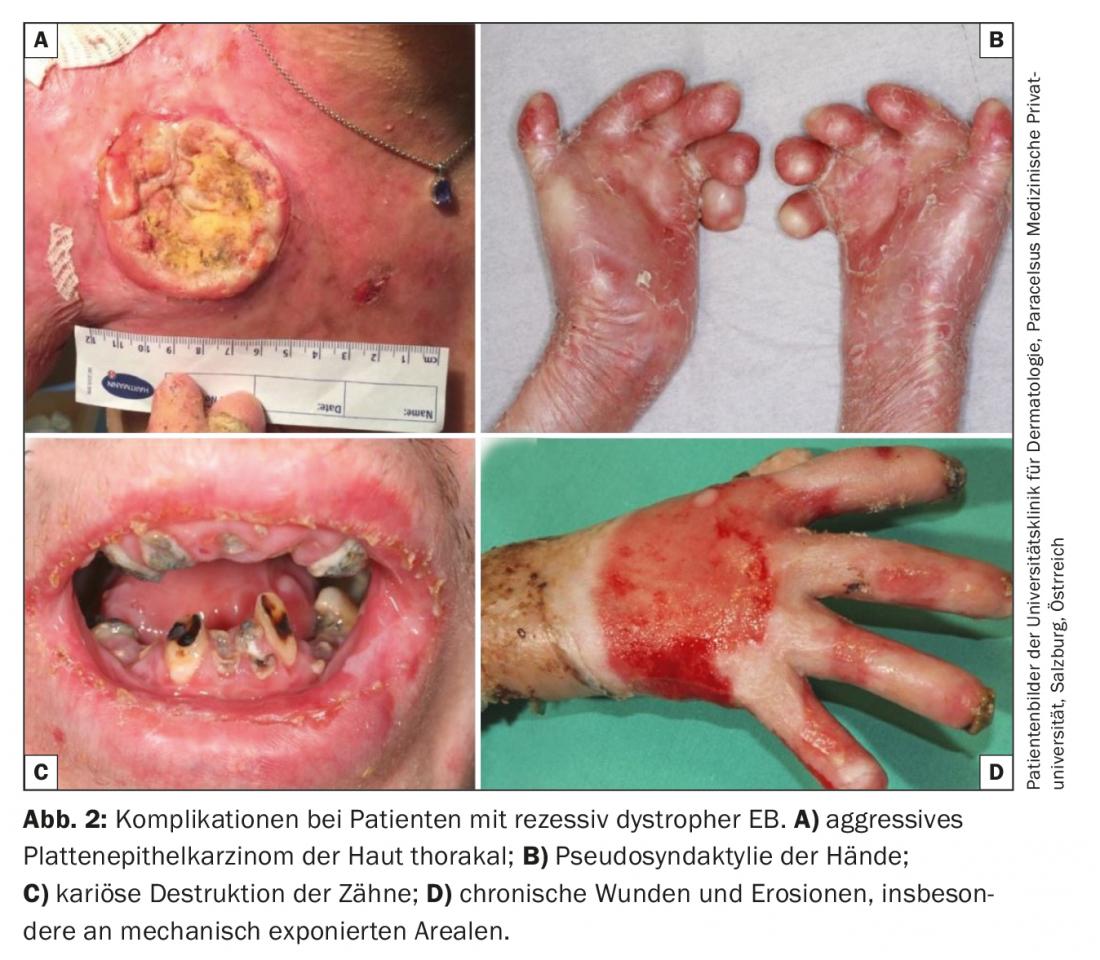
In assenza di opzioni terapeutiche curative nella pratica clinica, il focus della terapia per tutte le forme di EB è evitare i fattori di provocazione e ottimizzare gli approcci terapeutici sintomatici . Questi includono una terapia locale adeguata allo stadio della ferita, il miglioramento o il mantenimento della barriera cutanea attraverso una cura ottimizzata della pelle, il sollievo dal dolore e dal prurito, la prevenzione e il trattamento delle infezioni (terapia antisettica e antibiotica intermittente), il mantenimento di un apporto calorico e nutritivo adeguato e, infine, la prevenzione e il trattamento di complicazioni come l’anemia, l’osteoporosi e il carcinoma aggressivo a cellule squamose. (Fig. 2) [16,17].
Le prospettive di opzioni terapeutiche molecolari, potenzialmente anche curative, per l’EB sono incoraggianti in linea di principio, anche se la loro implementazione ampia, sicura, (durevolmente) efficiente e praticabile nella pratica clinica quotidiana per i pazienti affetti da EB è ancora difficile da valutare al momento. Lo spettro dei metodi è complesso:
Terapia genica: nel 2006, è stata eseguita per la prima volta una terapia genica con trapianto di cheratinociti corretti per il gene LAMB3 in un paziente con JEB autosomico recessivo (mutazione nel gene LAMB3) [18]. In questa terapia di ‘sostituzione genica’, le cellule cutanee coltivate sono state trasfettate con un vettore contenente una copia cDNA funzionale del gene mutante causativo e trapiantate sulle aree ferite come equivalenti della pelle epiteliale. La zona di giunzione dermo-epidermica è rimasta strutturalmente e funzionalmente stabile durante il periodo di follow-up di 14 anni fino ad oggi, senza alcuna evidenza di vesciche, infiammazione o reazione immunitaria al neoantigene introdotto terapeuticamente. In uno studio recente, i cheratinociti autologhi sono stati coltivati secondo lo stesso principio, le cellule staminali epidermiche sono state isolate e corrette geneticamente in modo specifico, espanse e poi trapiantate su ampie aree di ferita di un bambino di 7 anni con JEB (mutazione nel gene della laminina 332). In questo modo, è stato possibile ottenere una riepitelizzazione pari all’80% della superficie corporea. La risposta clinica è correlata all’espressione persistente della laminina 332 fino ad oggi [19]. L’efficacia è limitata dal numero (troppo) ridotto di cellule staminali epidermiche nelle colture primarie dei pazienti affetti da epidermolisi bollosa per garantire una correzione permanente, che si stanno esaurendo a causa delle ferite croniche e dell’avanzare dell’età, nonché dagli aspetti di sicurezza (per quanto riguarda i vettori virali e la genotossicità o la mutagenesi inserzionale indotta). Gli sviluppi tecnologici sono in corso di valutazione negli studi attuali e dovrebbero migliorare il profilo rischio-beneficio di questo metodo, che è particolarmente adatto per le ferite croniche circoscritte che sono altamente sintomatiche o a rischio di complicazioni a lungo termine [20].
Sono in fase di sviluppo anche gli approcci di “silenziamento genico” per le forme autosomiche dominanti di EB (cioè il silenziamento dell’allele mutante con “piccoli RNA interferenti”), nonché gli approcci di terapia genica topica di facile applicazione. In quest’ultimo caso, ad esempio, il vescicole extracellulari, che sono isolate da cellule staminali mesenchimali allogeniche e possono trasportare sia la proteina collagene di tipo 7 mancante che l’mRNA di COL7A1 alle cellule bersaglio [21, 22].
Le tecnologie di editing del genoma che utilizzano le proprietà delle nucleasi programmabili (ad esempio CRISPR/Cas9, TALEN, nucleasi a dito di zinco) si basano sulla modifica/correzione delle sequenze geniche mutate. In questo processo, viene indotta in modo specifico una rottura del doppio filamento nel DNA, le sequenze mutate vengono eliminate e vengono inserite sequenze estranee correttive, oppure singole basi vengono corrette dai meccanismi di replicazione cellulare. Tuttavia, le preoccupazioni sulla sicurezza, soprattutto per quanto riguarda la precisione insufficiente e i potenziali siti di legame fuori bersaglio, non giustificano ancora l’uso (in vivo) nell’uomo [23,24].
Un fenomeno clinico notevole, che può verificarsi in tutte le principali forme di EB, è la comparsa (spontanea) di aree cutanee sane senza vesciche, come risultato di eventi genetici correttivi, che si chiama mosaicismo revertente o “terapia genica naturale”. I tentativi di trapiantare i cheratinociti revertenti ottenuti negli studi clinici di fase I utilizzando biopsie di pugno sulle ferite EB colpite dopo l’espansione in vitro sono stati finora inferiori alle aspettative, soprattutto a causa della perdita progressiva delle cellule revertanti [25,26].
Terapia cellulare: i primi risultati del trapianto di cellule staminali del midollo osseo hanno mostrato cellule del donatore nella pelle (presumibilmente cellule staminali pluripotenti riprogrammate del midollo osseo) e una buona risposta, in parte per diversi anni, in alcuni pazienti con EB distrofica recessiva, nonostante la mancanza di prove di una concentrazione di collagene di tipo 7 ripristinata. Questo successo, il cui meccanismo sottostante non è ancora chiaro, è stato diminuito da un aumento significativo della mortalità periprocedurale. La revisione delle terapie di condizionamento e dei protocolli di trapianto dovrebbe migliorare la tolleranza [27,28].
I substrati delle terapie cellulari alternative includono le cellule stromali mesenchimali allogeniche applicate per via intradermica o endovenosa e le cellule staminali mesenchimali adipogeniche. Si è potuto osservare, almeno temporaneamente, un miglioramento della guarigione delle ferite e una riduzione dei segni infiammatori sulla pelle, che probabilmente è dovuta principalmente all’induzione di processi immunomodulatori favorevoli [29]. Un effetto clinico è già stato dimostrato negli studi iniziali con i fibroblasti autologhi wild-type o geneticamente corretti iniettati per via intradermica, che producono anche collagene di tipo 7 oltre ai cheratinociti [30,31].
Cellule simili alle cellule staminali embrionali pluripotenti possono anche essere derivate da cellule somatiche (ad esempio, fibroblasti o cheratinociti) mediante la trasfezione di tre o quattro fattori di trascrizione embrionali. L’uso di queste cosiddette cellule staminali pluripotenti indotte (iPSC), che possono differenziarsi nuovamente in vari tipi di cellule (ad esempio, cheratinociti o fibroblasti), viene studiato anche nell’EB negli studi preclinici. L’uso di cellule/cheratinociti revertanti per la produzione di iPSC ha un grande potenziale, in quanto si può fare a meno della correzione genica e dei rischi associati [32,33].
Terapie proteiche: La correzione della zona di giunzione dermo-epidermica difettosa viene tentata anche attraverso la sostituzione della proteina mancante o prodotta in modo difettoso. Mentre gli studi con l’applicazione di collagene ricombinante di tipo 7 sono ancora in fase preclinica, l’applicazione topica di un gel contenente un virus modificato di tipo 7 collagene che esprime l’herpes simplex di tipo 1, ad esempio, è già in fase di sperimentazione clinica [34,35].
Terapie basate sull’RNA/”piccole molecole”: Le mutazioni nonsense, che causano un codone di stop attraverso una mutazione puntiforme del DNA e quindi interrompono la traduzione, sono responsabili di circa il 10% di tutte le malattie genetiche umane. Farmaci come gli aminoglicosidi (ad esempio, la gentamicina) o l’immunomodulatore amlexanox possono portare a un “read-through” del codone di stop (ad esempio, della mutazione COL7A1) legandosi ai ribosomi in presenza di questa mutazione, permettendo così la produzione di proteine funzionali [36].
Le modifiche a livello dell’RNA si ottengono anche mediante il “salto dell’esone mediato da oligonucleotidi antisenso” (rimozione mirata degli esoni contenenti mutazioni) e il “trans-splicing dell’RNA mediato da spliceosomi” (SMaRT) (correzione dei segmenti di pre-RNA mutati). Quest’ultima tecnologia è già stata utilizzata in modo preclinico per correggere con successo una mutazione del gene della plectina nell’EB simplex e una mutazione COL7A1 nell’EB distrofica recessiva, nonché mutazioni autosomiche dominanti nel gene della cheratina-14 di una linea cellulare EB simplex [37,38].
Le cosiddette “piccole molecole” vengono utilizzate come mediatori di un effetto “modificante” la malattia. Questi includono il calcipotriolo topico, che si ritiene rafforzi le difese antimicrobiche endogene e migliori la guarigione delle ferite, aumentando l’espressione del peptide antimicrobico catelicidina. [39] e la diacereina topica, un componente della radice di rabarbaro e un potente inibitore dell’IL-1β proinfiammatorio, che ha dimostrato di ridurre la formazione di vesciche nei pazienti con EBS, secondo i dati preliminari pubblicati. [40]. L’inibitore orale della tirosin-chinasi rigosertib mostra anche un’inibizione selettiva delle cellule tumorali squamose dei pazienti con EB distrofica recessiva in vitro, e la sua efficacia e sicurezza sono in fase di test in uno studio in corso. Il carcinoma a cellule squamose altamente aggressivo come complicazione dell’epidemia cronica di EB Le ferite sono una delle principali cause di morte, in particolare nei pazienti con epidemia distrofica recessiva [41]. Oltre a rigosertib, l’anticorpo monoclonale anti-PD-1 nivolumab è attualmente in fase di sperimentazione controllata della sua efficacia nel carcinoma a cellule squamose localmente avanzato e metastatico nella coorte EB (EudraCT 2016-002811-16).
Messaggi da portare a casa
- L’epidermolisi bollosa (EB) è una malattia geneticamente e fenotipicamente eterogenea. I decorsi gravi si trasformano in una malattia multisistemica con morbilità e mortalità pronunciate. Le principali cause di morte sono infezioni, distrofia, insufficienza d’organo e carcinoma a cellule squamose. Queste ultime si manifestano precocemente e si moltiplicano nelle ferite croniche e hanno un decorso aggressivo.
- La diagnosi viene fatta in correlazione con la clinica, l’istologia, l’immunofluorescenza e l’analisi molecolare. Nonostante gli approcci innovativi, in parte causali, delle strategie di terapia molecolare, la cura è ancora una prospettiva futura indirettamente vaga. L’immunomodulazione, nel frattempo, è una strategia di trattamento promettente.
- I componenti epigenetici e biochimici secondari e i fattori ambientali, che possono indurre, ad esempio, cascate infiammatorie croniche e sistemiche, hanno un’elevata rilevanza patogenetica. Come per altre malattie rare, alcune caratteristiche rendono difficile condurre studi clinici e quindi generare prove di alta qualità.
Letteratura:
- Fine JD: Epidermolisi bollosa ereditaria. Orphanet J Rare Dis 2010; 5: 12.
- Fine JD, et al: Epidermolisi bollosa e rischio di tumori pericolosi per la vita: l’esperienza del Registro Nazionale EB, 1986-2006. J Am Acad Dermatol 2009; 60(2): 203-211.
- Fine JD, et al: La classificazione dell’epidermolisi bollosa ereditaria (EB): Relazione del Terzo Incontro Internazionale di Consenso sulla Diagnosi e la Classificazione dell’EB. J Am Acad Dermatol 2008; 58(6): 931-950.
- Bruckner-Tuderman L, et al: Progressi nella ricerca sull’epidermolisi bollosa: sintesi della Conferenza Internazionale di Ricerca DEBRA 2012. J Invest Dermatol 2013; 133(9): 2121-2126.
- Uitto J, Richard G: Progressi nell’epidermolisi bollosa: classificazione genetica e implicazioni cliniche. Am J Med Genet C Semin Med Genet 2004; 131C(1): 61-74.
- Kuttner V, et al.: Rimodellamento globale del microambiente cellulare a causa della perdita di collagene VII. Mol Syst Biol 2013; 9: 657.
- Odorisio T, et al.: I gemelli monozigoti discordanti per il fenotipo dell’epidermolisi bollosa distrofica recessiva evidenziano il ruolo della segnalazione TGF-beta nel modificare la gravità della malattia. Hum Mol Genet 2014; 23(15): 3907-3922.
- Pulkkinen L, et al: Nuove mutazioni ITGB4 nelle varianti letali e non letali dell’epidermolisi bollosa con atresia pilorica: missenso contro nonsenso. Am J Hum Genet 1998; 63(5): 1376-1387.
- Fine JD, et al: Epidermolisi bollosa ereditaria: raccomandazioni aggiornate su diagnosi e classificazione. J Am Acad Dermatol 2014; 70(6): 1103-1126.
- He Y, et al: Mutazioni monoalleliche nel codone di iniziazione della traduzione di KLHL24 causano fragilità cutanea. Am J Hum Genet 2016; 99(6): 1395-1404.
- Lee JYW, et al: Le mutazioni in KLHL24 si aggiungono all’eterogeneità molecolare dell’Epidermolisi Bullosa Simplex. J Invest Dermatol 2017; 137(6): 1378-1380.
- Lin Z, et al.: Mutazioni stabilizzanti dell’ubiquitina ligasi KLHL24 causano la perdita della cheratina 14 e la fragilità della pelle umana. Nat Genet 2016; 48(12): 1508-1516.
- Vahidnezhad H, et al: Una mutazione recessiva nella tetraspanina CD151 causa l’epidermolisi bollosa simile alla sindrome di Kindler con manifestazioni multisistemiche, tra cui la nefropatia. Matrix Biol 2018; 66: 22-33.
- Salo AM, et al: Un disturbo del tessuto connettivo causato da mutazioni del gene della lisil-idrossilasi 3. Am J Hum Genet 2008; 83(4): 495-503.
- Nischler E, et al: Insidie diagnostiche nei neonati e nei bambini con vesciche ed erosioni. Dermatol Res Practice 2009; 2009: 320403.
- El Hachem M, et al: Raccomandazioni di consenso multicentriche per la cura della pelle nell’epidermolisi bollosa ereditaria. Orphanet J Rare Dis 2014; 9: 76.
- Dănescu S, et al: Correlazione tra la gravità della malattia e la qualità della vita nei pazienti con epidermolisi bollosa. J Eur Acad Dermatol Venereol 2018. doi: 10.1111/jdv.15371. [Epub ahead of print]
- Mavilio F, et al: Correzione dell’epidermolisi bollosa giunzionale mediante trapianto di cellule staminali epidermiche geneticamente modificate. Nat Med 2006; 12(12): 1397-1402.
- Hirsch T, et al.: Rigenerazione dell’intera epidermide umana utilizzando cellule staminali transgeniche. Natura, 2017; 551(7680): 327-332.
- Siprashvili Z, et al: Sicurezza ed esiti delle ferite a seguito di innesti epidermici autologhi geneticamente corretti in pazienti con Epidermolisi Bullosa Distrofica Recessiva. JAMA 2016; 316(17): 1808-1817.
- Rosa J, et al: Gli attuali sistemi di consegna di siRNA non virali come trattamento promettente delle malattie della pelle. Curr Pharm Des 2018; 24(23): 2644-2663.
- McBride JD, et al: Doppio meccanismo di trasferimento del collagene di tipo VII da parte delle vescicole extracellulari delle cellule staminali mesenchimali del midollo osseo ai fibroblasti dell’epidermolisi bollosa distrofica recessiva. Biochimie 2018; 155: 50-58.
- Hainzl S, et al: Modifica di COL7A1 tramite CRISPR/Cas9 nell’Epidermolisi Bullosa Distrofica Recessiva. Mol Ther 2017; 25(11): 2573-2584.
- March OP, Reichelt J, Koller U: Editing genico per le malattie della pelle: le nucleasi di design come strumenti per la terapia genica dei disturbi della fragilità cutanea. Exp Physiol 2018; 103(4): 449-455.
- Gostynski A, Pasmooij AM, Jonkman MF: Trapianto terapeutico di successo di pelle revertita nell’epidermolisi bollosa. J Am Acad Dermatol 2014; 70(1): 98-101.
- van den Akker PC, et al.: Una “cellula revertente tardiva, ma che si adatta” spiega l’alta frequenza del mosaicismo revertante nell’epidermolisi bollosa. PLoS One 2018; 13(2): p. e0192994.
- Ebens CL, et al: Il trapianto di midollo osseo con ciclofosfamide post-trapianto per l’epidermolisi bullosa distrofica recessiva espande il pool di donatori affini e permette la tolleranza di innesti di cellule non ematopoietiche. Br J Dermatol 2019. doi: 10.1111/bjd.17858. [Epub ahead of print]
- Vanden Oever M, et al: Inside out: medicina rigenerativa per l’epidermolisi bollosa distrofica recessiva. Pediatr Res 2018; 83(1-2): 318-324.
- Ganier C, et al: L’iniezione intradermica di cellule stromali mesenchimali del midollo osseo corregge l’epidermolisi bollosa distrofica recessiva in un modello di xenotrapianto. J Invest Dermatol 2018; 138(11): 2483-2486.
- Petrof G, et al: La terapia cellulare con fibroblasti migliora la guarigione iniziale nelle ferite da epidermolisi bollosa distrofica recessiva: risultati di uno studio randomizzato, controllato con veicolo. Br J Dermatol 2013; 169(5): 1025-1033.
- Venugopal SS, et al: Uno studio randomizzato di fase II controllato con veicolo di fibroblasti allogenici intradermici per l’epidermolisi bollosa distrofica recessiva. J Am Acad Dermatol 2013; 69(6): 898-908.e7. doi: 10.1016/j.jaad.2013.08.014
- Itoh M, Kiuru M, Cairo MS, Christiano AM.: Generazione di cheratinociti da cellule staminali pluripotenti indotte da epidermolisi bullosa distrofica normale e recessiva. Proc Natl Acad Sci U S A 2011; 108(21): 8797-8802.
- Nakayama C, et al: Lo sviluppo di cellule staminali pluripotenti indotte derivate da cellule staminali mesenchimali/stromali da cheratinociti epidermici umani normali e RDEB. J Dermatol Sci 2018; 91(3): 301-310.
- South AP, Uitto J: Terapia sostitutiva con collagene di tipo VII nell’Epidermolisi Bullosa Distrofica Recessiva: quanto, quanto spesso? J Invest Dermatol 2016; 136(6): 1079-1081.
- NIH: Terapia genica topica con Bercolagene Telserpavec (KB103) per ripristinare il collagene VII funzionale per il trattamento dell’Epidermolisi Bullosa Distrofica (GEM-1), https://clinicaltrials.gov/ct2/show/NCT03536143, ultimo accesso 25 marzo 2019.
- Lincoln V, et al: La gentamicina induce il readthrough della mutazione nonsense LAMB3 e ripristina la funzionalità della laminina 332 nell’epidermolisi bollosa giunzionale. Proc Natl Acad Sci U S A, 2018; 115(28): E6536-E6545.
- Peking P, et al: Una strategia di trans-splicing dell’RNA non virale mediata da una pistola genetica per la riparazione di Col7a1. Mol Ther Nucleic Acids 2016. 5:e287. doi: 10.1038/mtna.2016.3.
- Turczynski S, et al: L’exon skipping mirato ripristina l’espressione del collagene di tipo VII e la formazione di fibre di ancoraggio in un modello di RDEB in vivo. J Invest Dermatol 2016; 136(12): 2387-2395.
- Guttmann-Gruber C, et al.: Il calcipotriolo a basso dosaggio può suscitare effetti di chiusura della ferita, antimicrobici e antineoplastici nei cheratinociti dell’epidermolisi bollosa. Sci Rep 2018; 8(1): 13430.
- Wally V, et al: Sviluppo del farmaco orfano Diacerein per l’epidermolisi bollosa simplex: uno studio clinico di fase 2/3 randomizzato, controllato con placebo e in doppio cieco. J Am Acad Dermatol 2018; 78(5): 892-901.e7. doi: 10.1016/j.jaad.2018.01.019.
- Atanasova VS, et al: Identificazione di rigosertib per il trattamento del carcinoma a cellule squamose associato all’epidermolisi bollosa distrofica recessiva. Clin Cancer Res, 2019: 10.1158/1078–0432.CCR-18–2661
PRATICA DERMATOLOGICA 2019; 29(2): 16-20