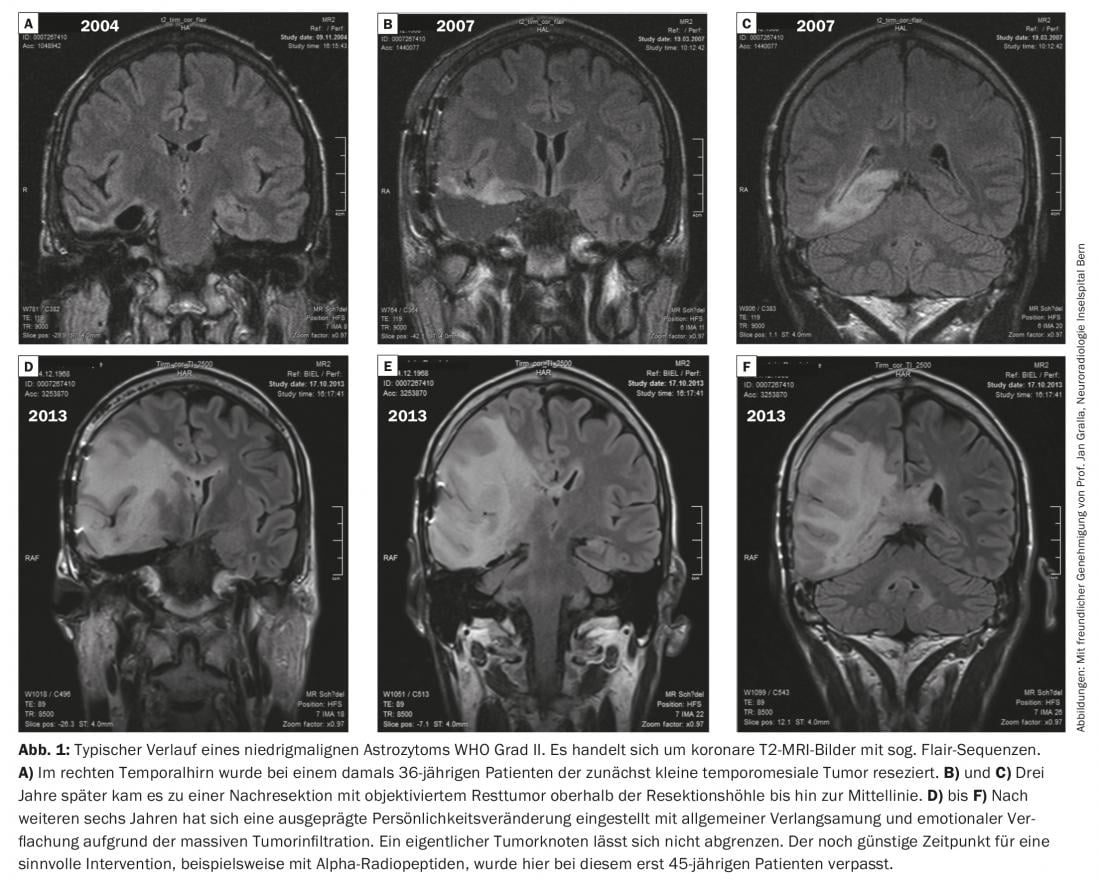
I gliomi di grado II tendono a essere sottovalutati nella loro malignità nella fase iniziale. Non esiste uno standard di trattamento. Oltre all’attesa, vengono sempre più spesso adottate tattiche chirurgiche aggressive. Tuttavia, anche in questo caso non è possibile ottenere una cura.
I gliomi poco maligni di grado OMS II spesso infiltrano la corteccia e quindi provocano crisi epilettiche nel 60-80% dei casi, spesso come manifestazione iniziale [1,2]. Il lobo più grande del cervello, il cervello frontale, è quello più spesso colpito. Si verificano cambiamenti di personalità, disturbi dell’impulso e dell’umore, ma anche atassia frontale. Se è colpita la regione fronto-precentrale e postcentrale, si verificano crisi motorie focali persistenti con tendenza alla generalizzazione, ma anche emiparesi spastica e disturbi sensoriali. Se è interessato il lobo temporale, predominano le crisi parziali complesse e persino i disturbi del linguaggio. I disturbi del sistema visivo sono rari; la causa più probabile è l’emianopsia omonima, quando è interessato il tractus opticus. Se viene colpito il tronco encefalico, si verificano complessi disturbi neurologici delle vie lunghe e della funzione dei nervi cranici, fino alla paralisi bulbare con disturbi della deglutizione e aspirazione. Se il talamo e i gangli della base sono colpiti, si verificano disturbi extrapiramidali o disturbi neurologici. Si evidenziano fluttuazioni nella vigilanza fino alla demenza. Spesso, tuttavia, la personalità è ancora ben conservata per molto tempo, anche nel caso di processi molto grandi, perché le cellule tumorali crescono diffusamente attraverso il tessuto cerebrale sano senza distruggerlo. Il lento spostamento delle aree funzionali da parte del tessuto tumorale consente uno spostamento della funzione minacciata nelle aree vicine o sul lato opposto (plasticità), che è riconoscibile nella risonanza magnetica funzionale.
La Figura 1 mostra il decorso tipico di un astrocitoma poco maligno di grado OMS II.
I gliomi: Manifestazione del tasso di mutazione spontanea?
Raramente (<5%), i tumori cerebrali si manifestano in modo sindromico nei tumori familiari, ad esempio nella sindrome di Turcot con difetto del gene “mismatch repair” o nella sindrome di Li-Fraumeni con mutazione di p53 [3–5]. Ancora più rari sono gli astrocitomi benigni a cellule giganti che si manifestano nell’infanzia e sporadicamente negli adulti come risultato di una mutazione congenita dei geni della sclerosi tuberosa TSC1 e TSC2, che co-regolano il complesso mTOR del metabolismo energetico. Il difetto genetico più comune associato alle malattie neurologiche è la mutazione del gene neurofibromatosi di tipo 1, con una frequenza di 1:3000. Si tratta di microdelezioni intrageniche, che nella metà dei casi non sono ereditate, ma si verificano spontaneamente. Il locus del gene NF1 sul cromosoma 17q11.2 è relativamente instabile. Il fenotipo Nf1 comprende i gliomi ottici benigni. Le delezioni di Nf1 sono state rilevate nei sottotipi di glioblastoma e contribuiscono alla genesi del glioma in combinazione con altre mutazioni. Le mutazioni dei geni dell’isocitrato deidrogenasi IDH1 e IDH2 si trovano frequentemente negli astrocitomi di grado II e nel glioblastoma secondario, prognosticamente migliore. Come la co-delezione 1p-19q nell’oligodendroglioma, la mutazione IDH segna un tipo di origine biologica diversa con una prognosi migliore.
Il tasso di mutazione spontanea è di 1:100.000 per divisione cellulare. Un organismo passa attraverso circa1014 divisioni cellulari fino alla completa differenziazione. Durante la vita, in ogni cellula si verificano innumerevoli errori di lettura durante la divisione cellulare – anche nel serbatoio delle cellule staminali – che vengono per lo più immediatamente corretti da meccanismi di riparazione intrinseci alla cellula. Tuttavia, ci sono sempre mutazioni che non vengono riconosciute o rilevate. non viene riparato, il che, in rari casi, contribuisce allo sviluppo di tumori. I gliomi appartengono alle cosiddette “malattie orfane”, alle malattie molto rare, con un’incidenza inferiore a cinque casi su 10.000 persone all’anno.
Classificazione genetica molecolare
I test genetici di recente sviluppo che scansionano l’intero genoma consentono una prognosi abbastanza accurata di tutti i tipi di glioma e permettono di assegnare correttamente i casi istologicamente poco chiari [6]. L’intero genoma viene analizzato alla ricerca di modelli di metilazione patologici. L’espressione di molti geni che controllano la proliferazione delle cellule tumorali e promuovono l’apoptosi è regolata dalle cosiddette isole CpG nella regione promotrice dei geni, che possono essere spente dalla metilazione, ad esempio nella differenziazione degli organi, ma anche nelle cellule tumorali. Un altro tipo di inattivazione genica è la delezione a lungo raggio di bracci cromosomici, che trasforma una sezione eterozigote in una sezione monozigote, spesso con reduplicazione. Si può ora creare un cosiddetto allelotipo su tutti i cromosomi, per registrare la perdita di eterozigosi. Per esempio, le monosomie 1p e 19q sono patognomiche per gli oligodendrogliomi a crescita lenta. Gli oligodendrogliomi di tipo selvaggio con eterozigosi 1p/19q hanno un comportamento molto più aggressivo. Nei glioblastomi, in genere si riscontra la monosomia del cromosoma 10 e la trisomia del cromosoma 7 con amplificazione dell’EGFR. L’analisi combinata di allelotipo e metiloma consente una classificazione molto precisa di tutti i gliomi, soprattutto nei casi in cui l’istologia solleva dubbi. Se negli studi sul glioblastoma si osservano i cosiddetti sopravvissuti a lungo termine, la diagnosi istologica deve essere verificata con il test molecolare, poiché il tasso di errore istologico negli studi su larga scala è fino al 7% [7].
Riduzione ritardata della massa alla manifestazione dei sintomi
Poiché la maggior parte dei pazienti con glioma di grado II si trova in buone condizioni cliniche al momento della diagnosi, spesso si sceglie una fase iniziale di osservazione [1,2]. Molti pazienti possono quindi condurre una vita normale con relativamente pochi sintomi per alcuni anni. Con questa strategia, si interviene solo se c’è stata una trasformazione in un livello di malignità superiore (grado III o IV) o se la malignità ha raggiunto un livello superiore (grado III o IV). se si manifestano sintomi neurologici fino a segni di pressione cerebrale (mal di testa, disturbi della vigilanza, nausea fino al vomito). Poi viene programmata la riduzione della massa, seguita da radiochemioterapia, o dimissioni.
Craniotomia da sveglio e neuromonitoraggio
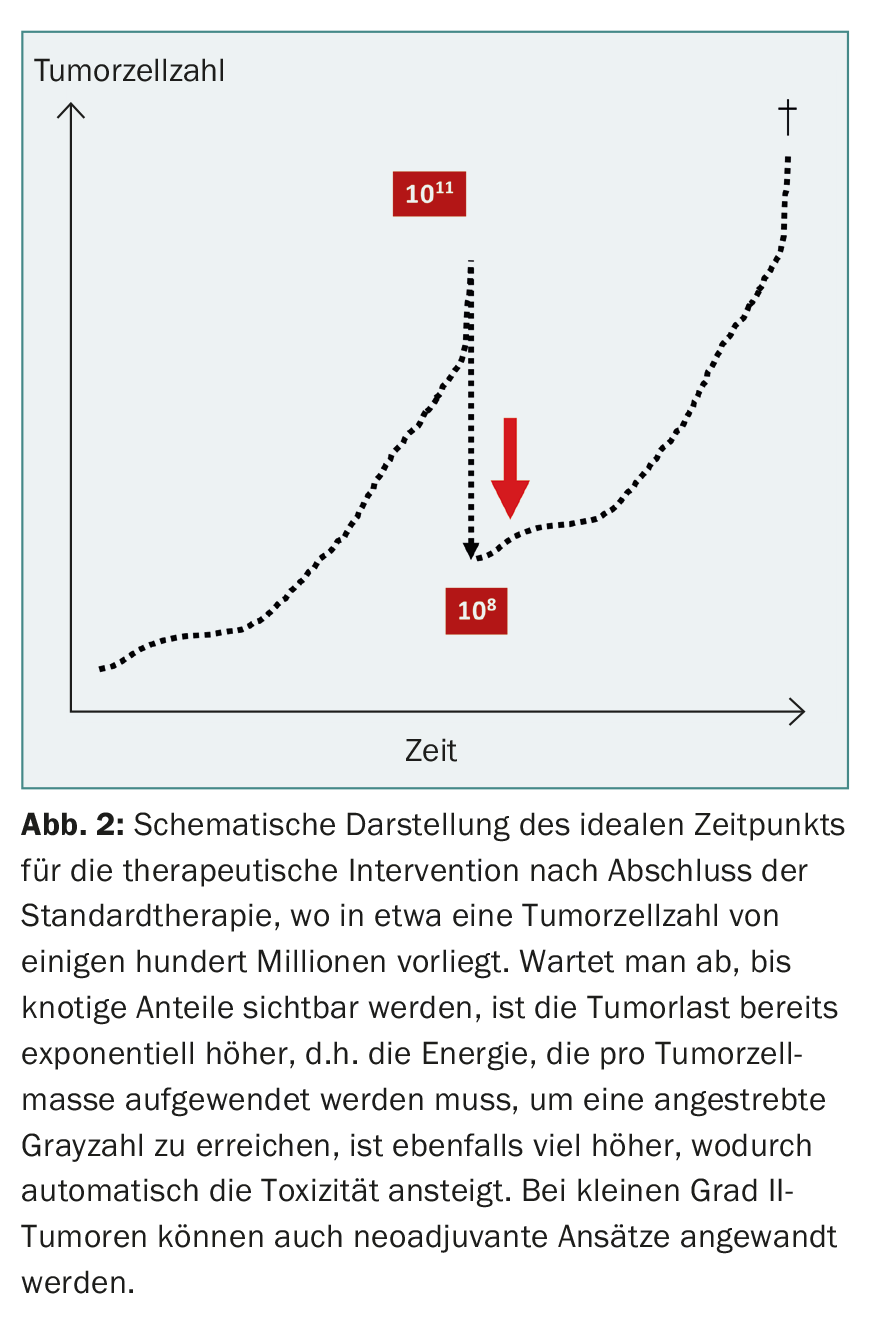
Nella fase iniziale della chirurgia del glioma, in casi disperati si è anche tentato di controllare il tumore con l’emisferectomia, che non poteva avere successo a causa della tendenza all’infiltrazione diffusa delle cellule tumorali. Negli ultimi anni, la chirurgia aggressiva del glioma ha conosciuto una rinascita, sebbene limitata alla riduzione estensiva della massa mediante craniotomia da svegli e neuromonitoraggio [8]. La riorganizzazione corticale dovuta alla plasticità cerebrale consente la resezione occasionale di aree importanti dal punto di vista funzionale, senza ulteriori deficit. Logicamente, questo approccio non può nemmeno portare a una cura, perché milioni di cellule tumorali infiltranti non vengono individuate. Questo può essere dimostrato con un modello di calcolo: Un corpo umano con un peso corporeo di 70 kg è composto da circa1014 cellule. Un tumore di 70 g contiene quindi circa 1011 cellule tumorali. Con una resezione del 99,9% di un tumore maligno infiltrante, rimangono circa 100 milioni di cellule tumorali, che determinano l’ulteriore destino (Fig. 2) . Una terapia moderna deve eliminare queste cellule residue o impedirne lo sviluppo. può controllare.
Prospettive terapeutiche
Molti pazienti affetti da astrocitoma di grado II non superano i 50 anni, oppure non vivono oltre i 50 anni. solo con deficit neurologici crescenti e sono esposti alla costante minaccia del 50% di probabilità di trasformazione in un livello di malignità superiore. Questo è un punto a favore dell’intervento precoce. La resezione sovramassimale non porta alla guarigione, ma riduce il rischio di trasformazione maligna. L’esperienza iniziale con l’irradiazione mirata di una singola cellula, utilizzando biomolecole diffusibili che si agganciano a recettori specifici delle cellule tumorali e trasportano un isotopo ad alta energia e a corto raggio come effettore, ha portato ripetutamente a un controllo del tumore molto prolungato, ben oltre la sopravvivenza mediana, senza tossicità significativa [9,10]. In futuro si aggiungeranno altri farmaci che bloccheranno i circuiti biologici regolatori disturbati, migliorando così la prognosi [5].
Messaggi da portare a casa
- Circa il 10-15% di tutti i gliomi maligni si presentano principalmente come astrocitomi di grado II a bassa malignità o, meno frequentemente, come oligodendrogliomi. La sopravvivenza mediana è da sette a dieci anni per gli astrocitomi di grado II e da dieci a 15 anni per gli oligodendrogliomi di grado II.
- Considerando la prognosi infelice dei glioblastomi più comuni, i gliomi di grado II tendono a essere sottovalutati nella loro malignità nella fase iniziale.
- I tumori cerebrali sono probabilmente una conseguenza del tasso di mutazione spontanea dei processi cellulari, in un certo senso il rovescio della medaglia della ricombinazione.
- La nuova classificazione OMS dei tumori cerebrali, basata sul modello di metilazione e sull’allelotipo, consente una dichiarazione affidabile sulla prognosi.
- Non esiste uno standard per il trattamento dei gliomi di grado II. Oltre all’approccio attendista, vengono sempre più spesso adottate tattiche chirurgiche aggressive. Tuttavia, a causa dell’infiltrazione delle cellule tumorali nel tessuto cerebrale sano, non è possibile ottenere una cura. Ci sono le prime indicazioni che la radioterapia mirata a singola cellula con biomolecole diffusibili potrebbe migliorare significativamente la prognosi.
Letteratura:
- Merlo A, de Tribolet N: Tumori del cervello e del midollo spinale. In: Steck A, Hess CH (eds.): Fisiopatologia neurologica. Berna: Verlag Hans Huber 2003.
- Schneider T, et al: Gliomi negli adulti. Dtsch Arztebl Int 2010; 107(45): 799-807.
- Merlo A, Rochlitz C, Scott R: Sopravvivenza dei pazienti con sindrome di Turcot e glioblastoma [letter]. N Engl J Med 1996; 334(11): 736-737.
- Merlo A, Bettler B: Glioblastomi in movimento. Scienza STKE 2004; 2004(229): pe18.
- Lino M, Merlo A: Tradurre la biologia in clinica: il caso del glioblastoma. Curr Opin Cell Biol 2009; 21(2): 311-316.
- Louis DN, et al: La classificazione dei tumori del sistema nervoso centrale dell’Organizzazione Mondiale della Sanità del 2016: una sintesi. Acta Neuropathol 2016; 131(6): 803-820.
- Linz U: Commento su Effetti della radioterapia con temozolomide concomitante e adiuvante rispetto alla sola radioterapia sulla sopravvivenza nel glioblastoma in uno studio randomizzato di fase III: analisi a 5 anni dello studio EORTC-NCIC (Lancet Oncol. 2009;10:459-466). Cancro 2010; 116(8): 1844-1846.
- Duffau H: Il razionale per eseguire una resezione precoce nel glioma diffuso di basso grado accidentale: verso una “neurooncologia chirurgica preventiva”. World Neurosurgery 2013; 80(5): e115-e117.
- Cordier D, et al: Terapia mirata con radionuclidi alfa dei gliomi in posizione funzionale critica con 213Bi-DOTAGA-sostanza P: uno studio pilota. Eur J Nucl Med Mol Imaging 2010; 37(7): 1335-1344.
- Cordier D, et al: Composti radiomarcati mirati nella terapia del glioma. Semin Nucl Med 2016; 46(3): 243-249.
InFo ONCOLOGIA & EMATOLOGIA 2017; 5(6): 7-10