La sclerosi sistemica (SSc) è una delle malattie più gravi perché è presente ovunque nel corpo, ma esiste una soluzione per pochi problemi e praticamente ogni organo deve essere trattato individualmente. Non esiste ancora un farmaco universale per loro, come per altre malattie, cioè non un biologico o un immunosoppressore specifico.
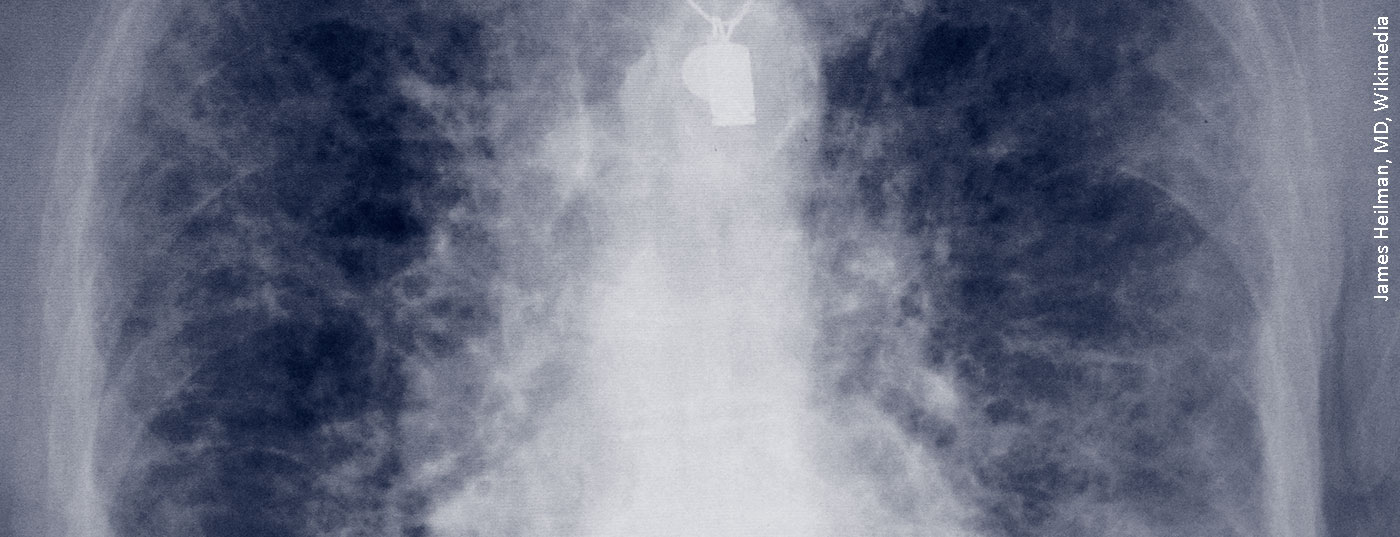
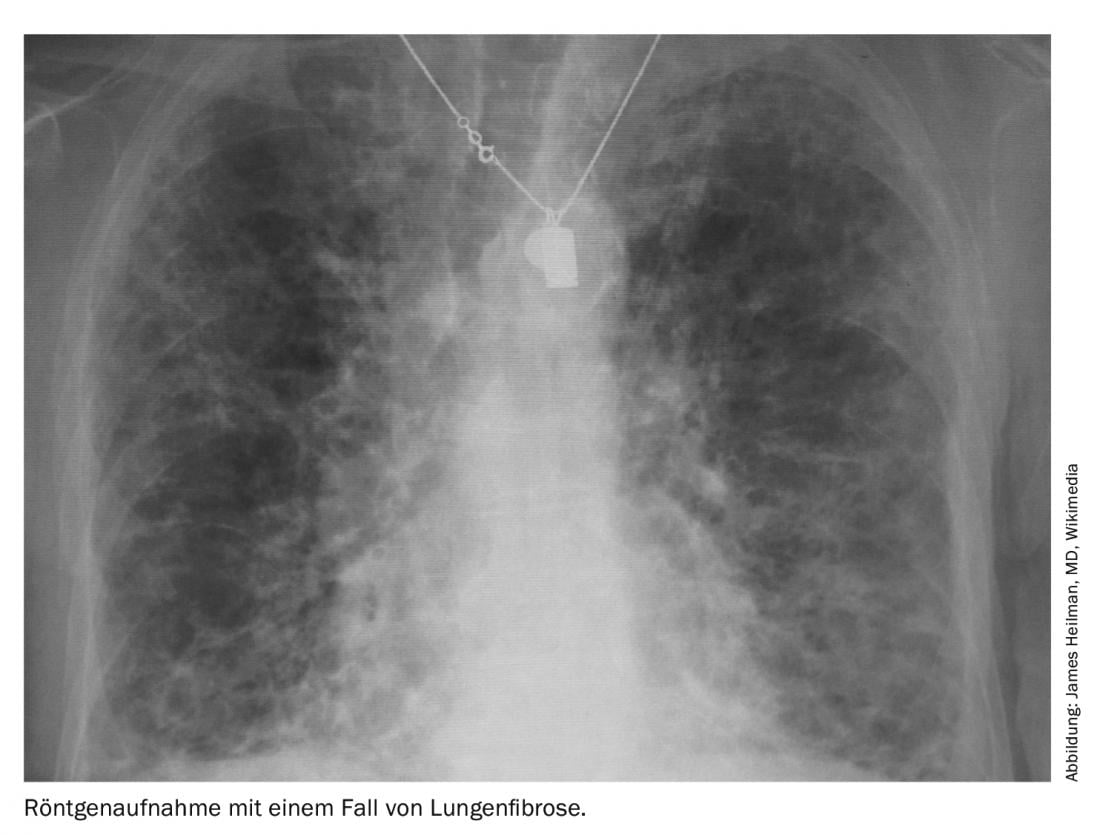
Un programma obbligatorio per ogni paziente che presenta una sclerosi sistemica è l’esame dei polmoni, per due componenti: una è la fibrosi polmonare, che è peggiore, e l’altra è l’ipertensione polmonare (PAH), per la quale almeno ci sono più farmaci. La perdita di estensibilità dei polmoni e la perdita della loro funzione di scambio di gas sono i due pato-meccanismi del modello di malattia della fibrosi polmonare. È 100 volte più rara, ma causa più decessi all’anno rispetto all’asma. Il dilemma è che ci sono più di cento condizioni diverse che possono portare allo stadio finale della fibrosi polmonare.
“Il deterioramento può essere molto rapido nella sclerosi sistemica, e questa è la parte pericolosa”, ha avvertito il Professor Ulf Müller-Ladner del Dipartimento di Reumatologia e Immunologia Clinica della Clinica Kerckhoff di Bad Nauheim (D). “I pazienti non hanno alcun segno importante, magari un’ulcera digitale, e improvvisamente la fibrosi polmonare o l’ipertensione polmonare sono già presenti”. I pazienti stessi se ne accorgono solo quando iniziano i sintomi della dispnea. Pertanto, secondo il reumatologo, è elementare nella sua disciplina esaminare sempre i polmoni di un paziente, indipendentemente dal fatto che presenti o meno dei sintomi.
Nella SSc, la fibrosi polmonare è diventata sempre più importante nell’elenco delle manifestazioni organiche. Anche perché altre complicazioni, come le crisi renali o i problemi cardiovascolari o gastrointestinali, sono diventate più trattabili nel tempo – la fibrosi polmonare associata alla SSc (SSc-ILD), invece, no. “Se si stratifica in base alle dimensioni sulla HRCT, i pazienti con SSc mostrano la fibrosi sulla TAC in oltre il 30% dei casi”, ha detto il Professor Dr Andreas Günther, Responsabile del Focus sulle malattie polmonari fibrose, Ospedale Universitario di Giessen Marburg. “Quindi è già una malattia più grave con un esito negativo”.
Migliore monitoraggio = risultato più controllato
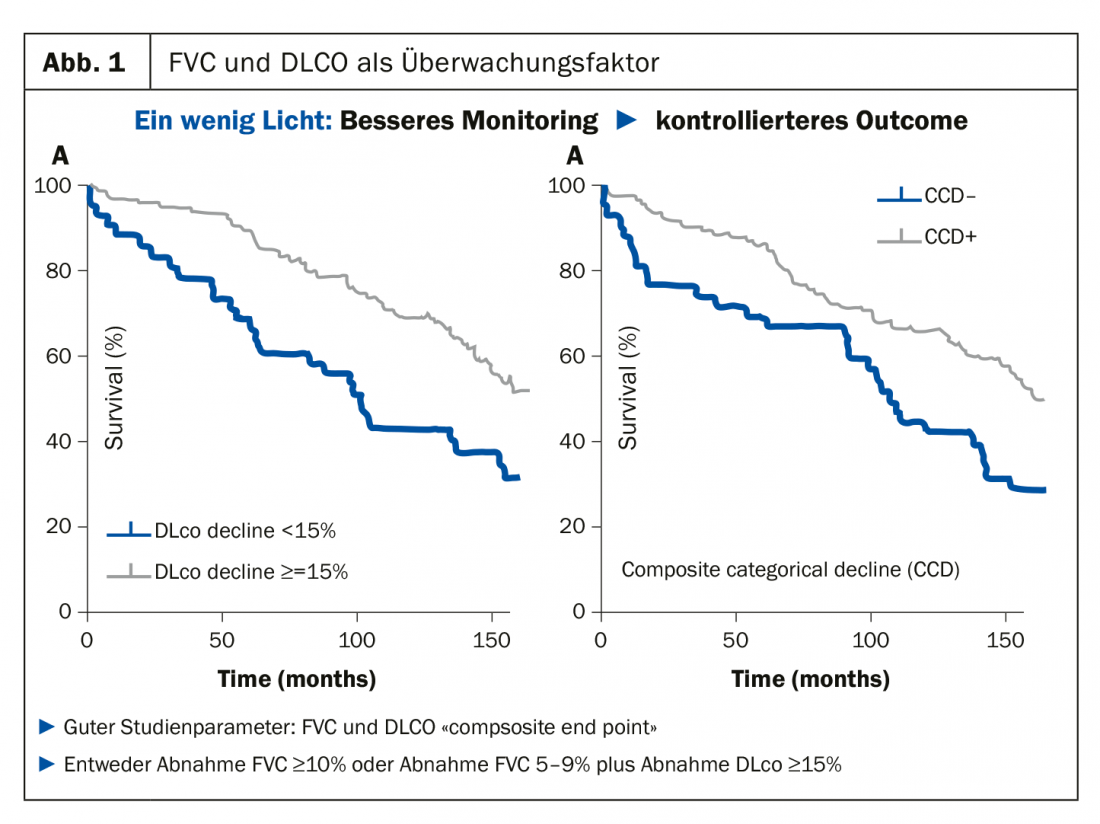
In qualità di reumatologo che deve tenere d’occhio e monitorare i polmoni, il Prof. Müller-Ladner consiglia sempre ai suoi colleghi di attenersi ai parametri relativamente facili da misurare, ossia la capacità vitale (FVC) e la capacità di diffusione (DLCO), poiché questa combinazione in particolare si è sviluppata molto positivamente come fattore di monitoraggio negli studi. “Se la combinazione di questi due parametri progredisce molto rapidamente, anche le curve divergono” (Fig. 1). Questo punto finale composito è ormai consolidato negli studi. Una diminuzione della FVC di ≥10% o una diminuzione del 5-9% più una diminuzione della DLCO di ≥15% devono essere considerate decisive. “Come reumatologi, possiamo sorridere di questi piccoli numeri, ma in relazione ai polmoni, sono assolutamente preziosi”, lo specialista ha chiarito un fattore che i suoi colleghi tendono a sottovalutare: “Un piccolo cambiamento nei polmoni può avere un grande impatto, e anche i pazienti lo notano”.
“La cosa principale è fare qualcosa”.
La terapia per molte malattie reumatologiche è simile: ci sono cellule infiammatorie, che vengono immobilizzate, e poi tutto va bene – gli effetti collaterali vengono lasciati da parte. La situazione è diversa con la SSc. “Perché qui ci sono diversi componenti che si muovono in parallelo e nessuno sa bene da cosa inizi – sistema immunitario, fibrosi, vascolare?”. E se abbassa uno di questi interruttori, potrebbe usarlo per alzare qualcosa altrove, il che non migliora la condizione generale. Allo stesso tempo, si ha a che fare con la fibrosi, “e antagonizzare la fibrosi è uno dei compiti più difficili in assoluto”. Rimane quindi la domanda su come procedere nel trattamento: togliere l’innesco o fare un legame diretto?
I reumatologi rispondono a questa domanda facendo ciò che possono e sanno, secondo il Prof. Müller-Ladner: assumono metotrexato o penicillamina, “la cosa principale è fare qualcosa”. Ma a volte l’azionismo può anche causare danni, ad esempio se la diagnosi non è corretta. Se i risultati non sono chiari, l’esperto dice che bisogna sempre consultare uno pneumologo per essere sicuri. Il Prof. Günther ha fatto riferimento alle linee guida di trattamento EULAR/EUSTAR SSc-ILD del 2016, che raccomandano la ciclofosfamide o il trapianto autologo di cellule staminali ematopoietiche (HSCT) (Tab. 1). Il micofenolato mofetile (MMF), secondo lo pneumologo, non è certamente meno efficace della ciclofosfamide, ma meno tossico, motivo per cui viene ancora spesso prescritto.

Studi promettenti
Un altro problema: la sclerosi sistemica è e rimane una malattia rara, che rende desiderabili ma scarsi gli studi di grandi dimensioni. “Scuotere il polso come si fa con l’artrite reumatoide (RA) non funziona perché non si riesce a trovare i numeri”, ha spiegato il Prof. Müller-Ladner. Ecco perché ogni pubblicazione è preziosa. Il reumatologo ha presentato prima due studi (FaSScinate e FocuSSced) con un numero relativamente piccolo di partecipanti, entrambi incentrati sulla lotta all’infiammazione con l’interleuchina-6 (IL-6) mediante tocilizumab. Soprattutto nello studio FocuSSced, si è visto che nell’intervallo a lungo termine, l’inibizione dell’infiammazione legata alla FVC sotto IL-6 era promettente.
Lo studio SENSCIS, a sua volta, ha analizzato l’effetto di nintedanib rispetto al placebo con l’endpoint primario della riduzione della capacità vitale funzionale (mL/anno). È orientato verso una medicazione di base stabile e adattata alla vita reale con MTX o MMF. Anche in questo caso, i risultati sono stati positivi dopo 52 settimane (Fig. 2). “Abbiamo bisogno di un farmaco antifibrotico sensato”, confida il Prof. Müller-Ladner, “perché finora la fibrosi può essere trattata solo indirettamente”.
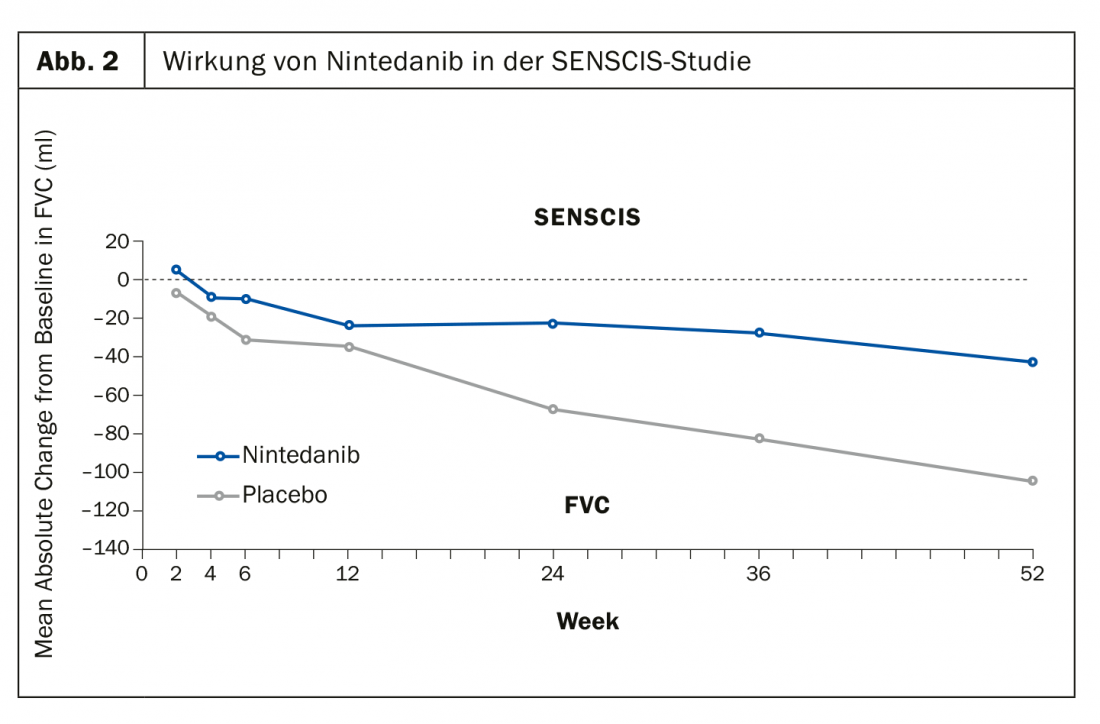
Tuttavia, nintedanib non è attualmente approvato per il trattamento della SSc/SSc-ILD o di altre malattie polmonari interstiziali progressive (PF-ILD) al di fuori della fibrosi polmonare idiopatica (IPF). Ciò rende ancora più importante ottenere l’approvazione nel prossimo futuro non solo per le forme fibrotiche puramente polmonari della malattia.
Fonte: Simposio industriale “Fibrosi polmonare: la sfida della SSc e di altre malattie reumatiche” nell’ambito della DGRh; organizzatore: Boehringer Ingelheim
InFo PNEUMOLOGIA & ALLERGOLOGIA 2019; 3(1): 30-31 (pubblicato l’11 dicembre 19, prima della stampa).