
Il 55° congresso annuale della Società Americana di Ematologia si è svolto alla fine del 2013. Oltre 22.000 partecipanti sono giunti a New Orleans, o “The Big Easy”, come viene chiamata la città. Gli interessati possono trovare il programma completo con gli estratti delle presentazioni su www.hematology.org.
Trapianto di cellule staminali aploidentiche
(mb) Il trapianto di cellule staminali ematopoietiche aploidentiche HLA è un’opzione efficace per i pazienti che hanno bisogno di un trapianto ma non riescono a trovare un donatore con tipi HLA completamente corrispondenti. Questo è il riassunto di Alice Bertaina, MD, Ospedale Pediatrico Bambino Gesù, Roma. Questo tipo di trattamento è solitamente associato a un rischio più elevato di infezione e di ricaduta rispetto a un trapianto di cellule staminali da un donatore completamente compatibile. Nel metodo mostrato da Bertaina, le cellule T alfa/beta+ e le cellule B CD19+ vengono rimosse selettivamente dall’innesto del donatore. Allo stesso tempo, si conservano le cellule immunoattive come le cellule natural killer e le cellule T gamma/delta+. Un totale di 45 bambini con leucemia acuta – di età compresa tra 0,9 e 17,9 anni al momento del trapianto – sono stati trattati con cellule staminali di un genitore (35 pazienti con ALL e 10 con AML). Il tempo medio per raggiungere una conta assoluta dei neutrofili di >0,5 × 109/l e una conta piastrinica di >50 × 109/l è stato rispettivamente di 13 (9-18) e 11 giorni (8-20). Nessun bambino ha sviluppato una reazione acuta trapianto-ospite (GVHD) nell’intestino o nel fegato. La GVHD lieve si è verificata nella pelle del 29%. Dopo un follow-up medio di undici mesi (tra 2 e 30), la stima di Kaplan-Meier a due anni era un tasso di sopravvivenza libera da leucemia del 75% (95% CI 57-86). Questo valore era del 73% (95% CI 52-85) per i pazienti ALL. “I nostri risultati con le cellule staminali da donatore aploidentico hanno mostrato percentuali di successo coerenti con quelle di un trapianto full-match”, ha spiegato il dottor Bertaina. “Ciò renderà disponibile questo trattamento salvavita a un numero molto maggiore di pazienti che non hanno un donatore perfetto”.
Recettore antigenico chimerico (CAR)
Diversi ricercatori hanno presentato un altro tour de force tecnico: una terapia con linfociti T modificati CAR per le malattie oncoematologiche. Il recettore dell’antigene chimerico (CAR) è una catena di DNA costruita artificialmente che viene inserita nelle cellule T di un paziente mediante vettori (noti dalla genetica molecolare). Queste ‘nuove’ cellule combinano la specificità degli anticorpi con l’effetto citotossico delle cellule T.
Michael Kalos della University of Pennsylvania Perelman School of Medicine di Filadelfia ha presentato i risultati di studi condotti sulla malattia CLL e ALL in fase avanzata, recidivata o refrattaria (Porter et al NEJM 2011; Kalos et al Sci Trans Med 2011, Grupp et al NEJM 2013, abstract 67). Sono stati trattati in totale 32 pazienti con CLL pesantemente pretrattati, con otto pazienti che hanno avuto una remissione parziale e sette una remissione completa. I risultati sono riportati nella Tabella 1.
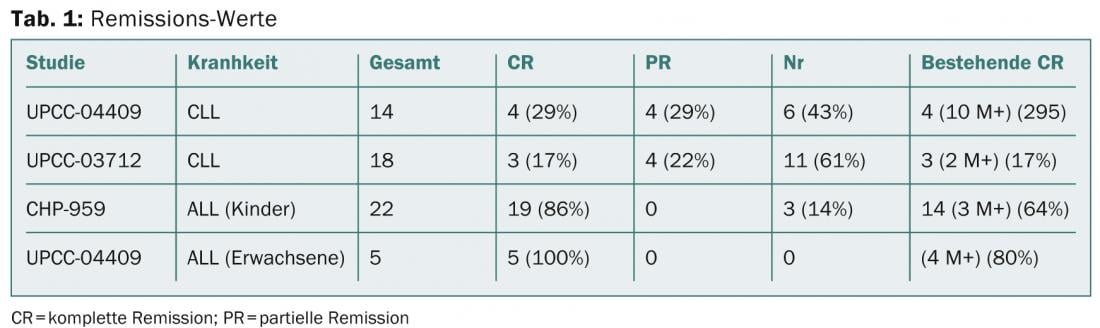
CAR per il linfoma a cellule B
Il fatto che anche i pazienti con linfoma primario a cellule B mediastiniche (PMBCL) e linfoma diffuso a grandi cellule B (DLBCL) refrattario alla chemioterapia possano trarre beneficio dalla terapia CAR anti-CD19 è dimostrato nel rapporto di James Kochenderfer, MD, del National Cancer Institute del National Institute of Health a Bethesda. I ricercatori hanno trattato 20 pazienti con un totale di 23 infusioni di cellule T. Sono già stati riportati i primi nove trattamenti con cellule CAR-T (Kochenderfer et al. in Blood 2010 e Blood 2012). All’ASH, Kochenderfer ha presentato i risultati non ancora comunicati di 14 pazienti. Poiché la precedente chemioterapia ha chiarito che l’attività delle cellule T trasferite adottivamente migliora, i pazienti ricevono ciclofosfamide più fludarabina per cinque giorni per un’infusione di cellule T CAR anti-CD19. Cinque pazienti hanno ottenuto una remissione completa (CR) e sei una remissione parziale (PR).
“Questo è il primo rapporto di successo del trattamento di PMBCL e DLBCL refrattari alla chemioterapia con cellule T CAR anti-CD19”, ha detto Kochendorfer. “I nostri dati sono i primi a suggerire il potenziale di questo approccio per i pazienti con linfomi aggressivi che sono stati quasi non trattabili”.
Zevalin e rituximab a confronto
Le terapie di consolidamento con Zevalin (90Y ibritumomab-tiuxetan) e rituximab hanno entrambe mostrato un beneficio significativo sulla sopravvivenza libera da progressione (PFS). Uno studio spagnolo di fase II ha chiarito che la terapia di consolidamento con rituximab è superiore al consolidamento con Zevalin nei pazienti con linfoma follicolare che rispondono a R-CHOP. Da giugno 2008 a luglio 2010, sono stati inclusi 146 pazienti (66 uomini e 80 donne con un’età media di 55 anni) provenienti da 25 istituti spagnoli. Armando Lopez-Guillermo, MD, dell’Hospital Clinic de Barcelona, ha presentato i risultati preliminari. Dopo un follow-up medio di 37 mesi dalla randomizzazione (tra 26 e 56), 31 pazienti hanno mostrato una progressione avanzata o una recidiva. La PFS a 36 mesi è stata del 64% (95% CI 52-76) per i pazienti del gruppo Zevalin e dell’86% (95% CI 77-95) per i pazienti del gruppo rituximab (p=0,01; HR 0,38, 95% CI 0,170,83). Durante il periodo di mantenimento, si è verificata neutropenia (grado 3-4) in sei dei 63 pazienti del gruppo Zevalin e trombocitopenia (3-4) in cinque di loro. Per i pazienti del gruppo rituximab, si trattava di uno o nessuno dei 61 pazienti. Cinque pazienti sono morti durante il follow-up a causa di un linfoma progressivo. Non è stata riscontrata alcuna differenza tra i gruppi.
Bortezomib nel mieloma multiplo
Nel mieloma multiplo (MM), il trattamento di induzione e mantenimento con bortezomib continua a migliorare la sopravvivenza dei pazienti sintomatici di nuova diagnosi. Questo è il risultato dello studio hOVON65, i cui risultati a lungo termine sono stati presentati da Pieter Sonneveld, MD, dell’Erasmus Medisch Centrum, Rotterdam. In questo studio, 827 pazienti con MM sono stati randomizzati a VAD (vincristina, doxorubicina, desametasone; n=414) o PAD (bortezomib, doxorubicina, desametasone; n=413) dopo la terapia di induzione, seguita da melfalan ad alte dosi e trapianto di cellule staminali autologhe. La terapia di mantenimento consisteva in 50 mg di talidomide al giorno (per il gruppo VAD) o 1,3 mg di bortezomib/m2 iv bisettimanale (per il gruppo PAD) per due anni. La risposta durante il trattamento di protocollo è sembrata leggermente migliorata ora che tutti i pazienti hanno completato il trattamento: La remissione completa (CR) più la remissione completa nodulare (nCR) si sono verificate nel 49% con PAD e nel 35% con VAD. La remissione parziale molto buona (VGPR) è stata osservata nel 26% e nel 21%, e la remissione parziale ≥ (PR) nel 91% e nell’83%, rispettivamente. Dopo un follow-up medio di 67 mesi, 111 dei pazienti trattati con VAD e 131 di quelli trattati con PAD erano liberi da progressione e vivi. La sopravvivenza libera da progressione (il tempo trascorso dalla randomizzazione alla progressione, alla recidiva o alla morte) è stata migliore con la PAD (aggiustata per lo stadio) (ISS) (HR 0,78; 95% CI 0,66-0,91; p=0,002). Il PAD era superiore anche per l’esito secondario della sopravvivenza globale dopo l’aggiustamento ISS (HR 0,80; 95% CI 0,65-1,00; p=0,047).
Nuova mutazione nella JAK-2-negativa e nella PMF
Ricercatori austriaci e italiani hanno scoperto una nuova mutazione specifica per i pazienti con trombocitemia essenziale (ET) JAK-2-negativa e mielofibrosi primaria (PMF). L’alterazione genetica più comune nella sindrome mieloproliferativa (MPN) è la mutazione JAK2-V617F. Questo si verifica nel 95% dei pazienti con policitemia vera (PV) e nel 50-60% dei pazienti con ET e PMF. Le mutazioni nell’esone 12 di JAK2 e nel gene del recettore della trombopoietina MPL sono presenti in un ulteriore 5-10% dei casi. Negli ultimi anni, è emerso che diversi altri geni sono interessati dalla MPN. Tuttavia, queste mutazioni si trovano anche in altre malattie mieloidi. Un marcatore molecolare specifico per il restante 40% dei pazienti con ET e PMF con JAK2 wild-type e MPL è quindi molto gradito.
Thorsten Klampfl del Centro di Ricerca per la Medicina Molecolare dell’Accademia Austriaca delle Scienze di Vienna e i suoi colleghi hanno identificato nuove mutazioni nei pazienti con PMF con JAK2 e MPL di tipo selvatico, utilizzando il sequenziamento dell’intero esoma. L’analisi ha mostrato inserzioni e delezioni somatiche ricorrenti nel CALR che codifica per la calreticulina. Tutte le mutazioni rilevate derivavano da uno spostamento della cornice di lettura ed erano raggruppate nell’ultimo esone (esone 9) del gene. In seguito a questa scoperta, i ricercatori hanno sviluppato un test basato sulla PCR che hanno utilizzato per esaminare 1.107 pazienti MPN per le mutazioni di inserzione/delezione nell’esone 9 di CALR. Non sono state trovate mutazioni nei pazienti PV, ma sono state trovate nei pazienti ET e PMF: le mutazioni CALR erano mutuamente esclusive con JAK2 mutato e MPL mutato. Tra i pazienti con JAK2 e MPL di tipo selvaggio, il 67% dei pazienti con ET e l’88% dei pazienti con PMF avevano CALR mutante. Non sono state rilevate mutazioni dell’esone 9 di CALR in 254 pazienti con AML de novo, in 45 con leucemia mieloide cronica, in 73 con sindrome mielodisplastica o in 64 con leucemia mieloide cronica.
Gli autori prevedono che il marcatore sarà presto a disposizione delle cliniche per migliorare le decisioni diagnostiche e terapeutiche nella MPN.
Fonte: 55° Meeting annuale ASH, 7-10 dicembre 2013, New Orleans
InFo Oncologia & Ematologia 2014; 2(3): 30-32
Speciale Congresso 2014; 5(2): 6-7









