La malattia autoimmune sistemica APS (sindrome antifosfolipidica) è caratterizzata principalmente da manifestazioni trombotiche. Come trombofilia acquisita, è indicata la tromboprofilassi con farmaci variabili.
La sindrome antifosfolipidica (APS) è una malattia autoimmune sistemica caratterizzata dal verificarsi di trombosi venose e/o arteriose e/o complicazioni della gravidanza [1]. La patogenesi dell’APS si basa sulla presenza di anticorpi diretti contro una varietà di complessi fosfolipidi/proteine [2]. La scoperta e la conoscenza di questi componenti abbraccia diversi decenni (Tab. 1).

Il pato-meccanismo di come gli anticorpi antifosfolipidi (aPL) portano all’attivazione della coagulazione è multifattoriale (Tab. 2). Esistono diversi approcci di studi sull’uomo e sugli animali che dimostrano il coinvolgimento di aPL [2]. L’APS può manifestarsi in modo isolato (APS primaria) o nel contesto di altre malattie autoimmuni (ad esempio, LES, tumori maligni). Una forma rara ma molto grave di APS è la sindrome antifosfolipidica catastrofica con coinvolgimento simultaneo di tre o più sistemi di organi [3].

Anticorpi antifosfolipidi (aPL)
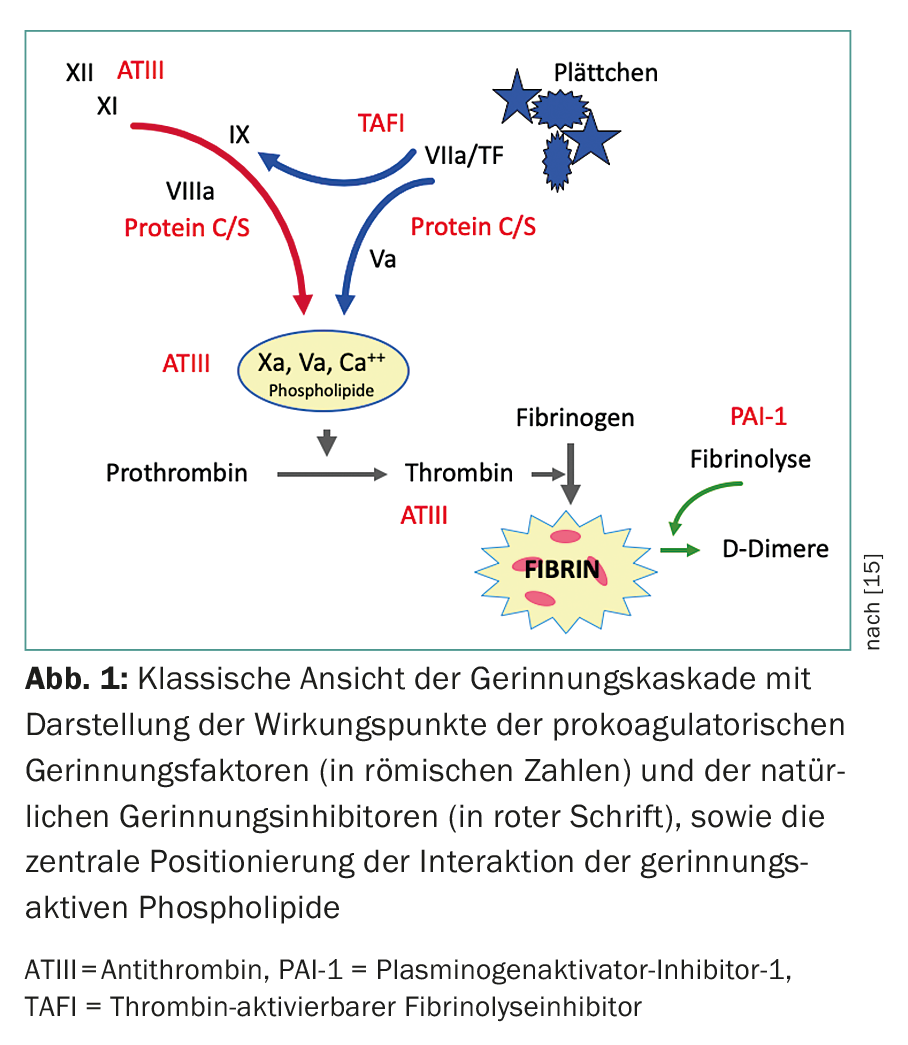
Gli anticorpi antifosfolipidi sono un gruppo eterogeneo di anticorpi acquisiti che sono diretti contro i complessi fosfolipidi-proteine e mostrano un’elevata affinità per le superfici cariche negativamente. Gli antigeni target (proteine) più importanti includono la β2-glicoproteina-I (β2-GPI) e la protrombina. Sono stati descritti anche altri antigeni come la proteina C attivata, la proteina S, l’annexina V, le LDL ossidate e il fattore XII ( Fig. 1) [2,4]. Le interazioni di aPL sulle superfici fosfolipidiche caricate negativamente provocano varie reazioni, come l’inibizione della regolazione emostatica (tramite l’inibizione della proteina C o dell’antitrombina, o lo spostamento dell’annessina V dai tromboblasti nella placenta), l’attivazione delle piastrine (tramite la stimolazione del trombossano A2), l’aumento della regolazione delle molecole di adesione e del fattore tissutale sulle cellule endoteliali e l’attivazione del complemento. (Tab.2).
Gli aPL possono causare un prolungamento del tempo di coagulazione nei test di coagulazione dipendenti dai fosfolipidi (ad esempio, Quick/INR o APTT), in quanto interferiscono o bloccano i fosfolipidi del complesso della protrombinasi, necessari per il decorso della cascata coagulativa. Questo fenomeno in vitro ha portato al nome fuorviante di “anticoagulante del lupus” nel 1952, quando questo effetto “anticoagulante” è stato osservato nei pazienti con lupus eritematoso nei test di laboratorio.
Criteri di classificazione
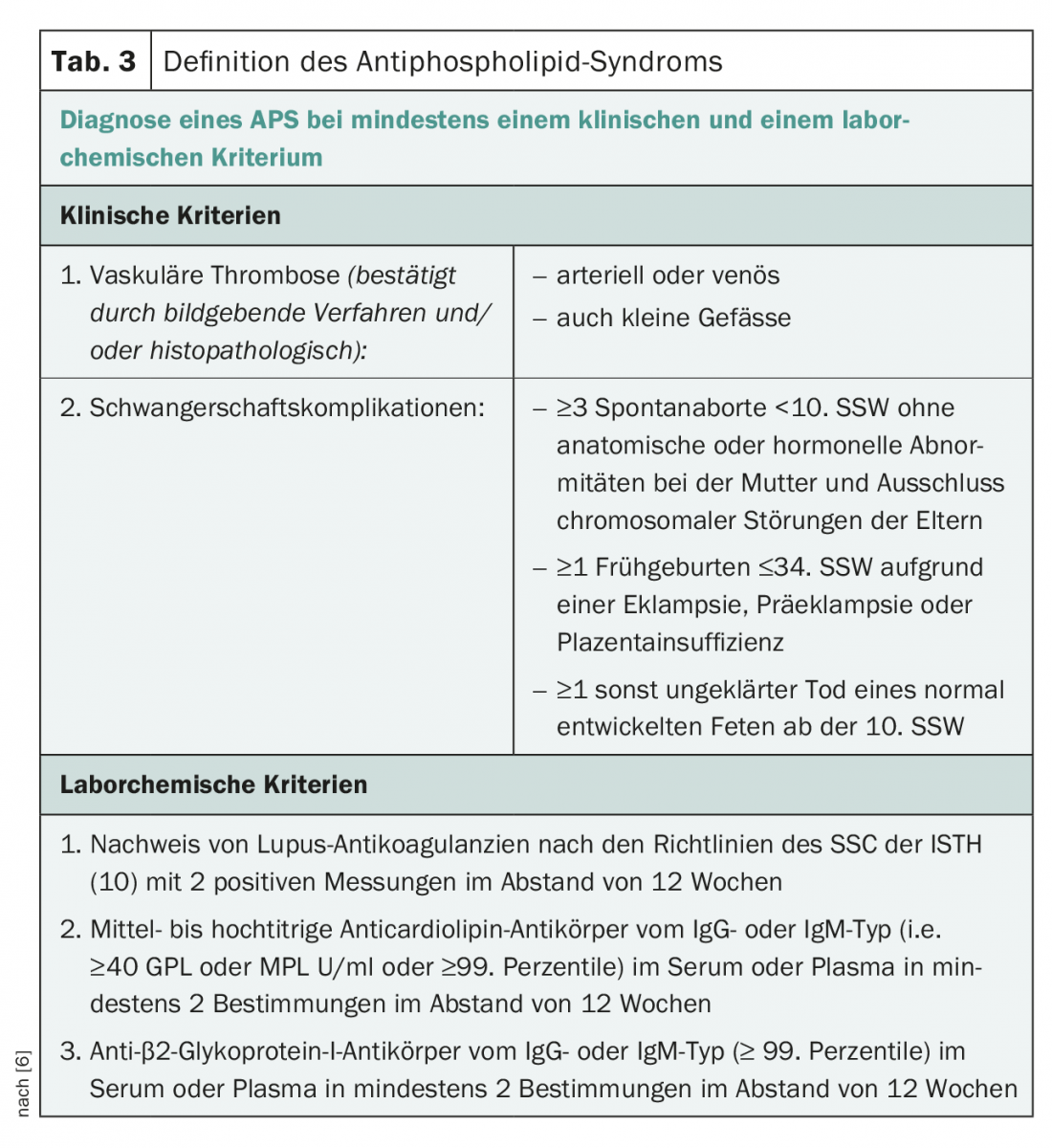
I criteri di classificazione dell’SPG sono stati formulati per la prima volta durante un workshop a Sapporo (Giappone) nel 1998 e rivisti nel 2005 in occasione dell'”11° Congresso Internazionale sugli Anticorpi Antifosfolipidi” a Sydney [5,6]. Pertanto, un criterio clinico (trombosi venosa e/o arteriosa nel micro o macrocircolo e complicazioni ostetriche) più un criterio di laboratorio (due positivi entro 12 settimane per il lupus anticoagulante, l’anticorpo anticardiolipina o l’anticorpo anti-β2GPI) sono necessari per definire l’APS definitiva (tab. 3).
Diagnostica dell’SPG
Per il rilevamento di aPL (LA, aCL-IgG /-IgM, β2GPI-IgG/-IgM), vengono combinati diversi metodi di test secondo le raccomandazioni internazionali [7,8]. Fondamentalmente, gli anticoagulanti del lupus sono identificati funzionalmente dai test di coagulazione. Gli anticorpi aCL e β2GPI, invece, vengono rilevati immunologicamente, mediante ELISA o saggio di chemiluminescenza. La determinazione di aPL due volte a distanza di almeno dodici settimane l’una dall’altra è necessaria per evitare il rilevamento di eventuali anticorpi transitori che possono verificarsi, ad esempio, nel contesto di infezioni. La Figura 2 illustra l’algoritmo diagnostico, che ha lo scopo di illustrare la complessità delle fasi diagnostiche nei casi di sospetta APS.
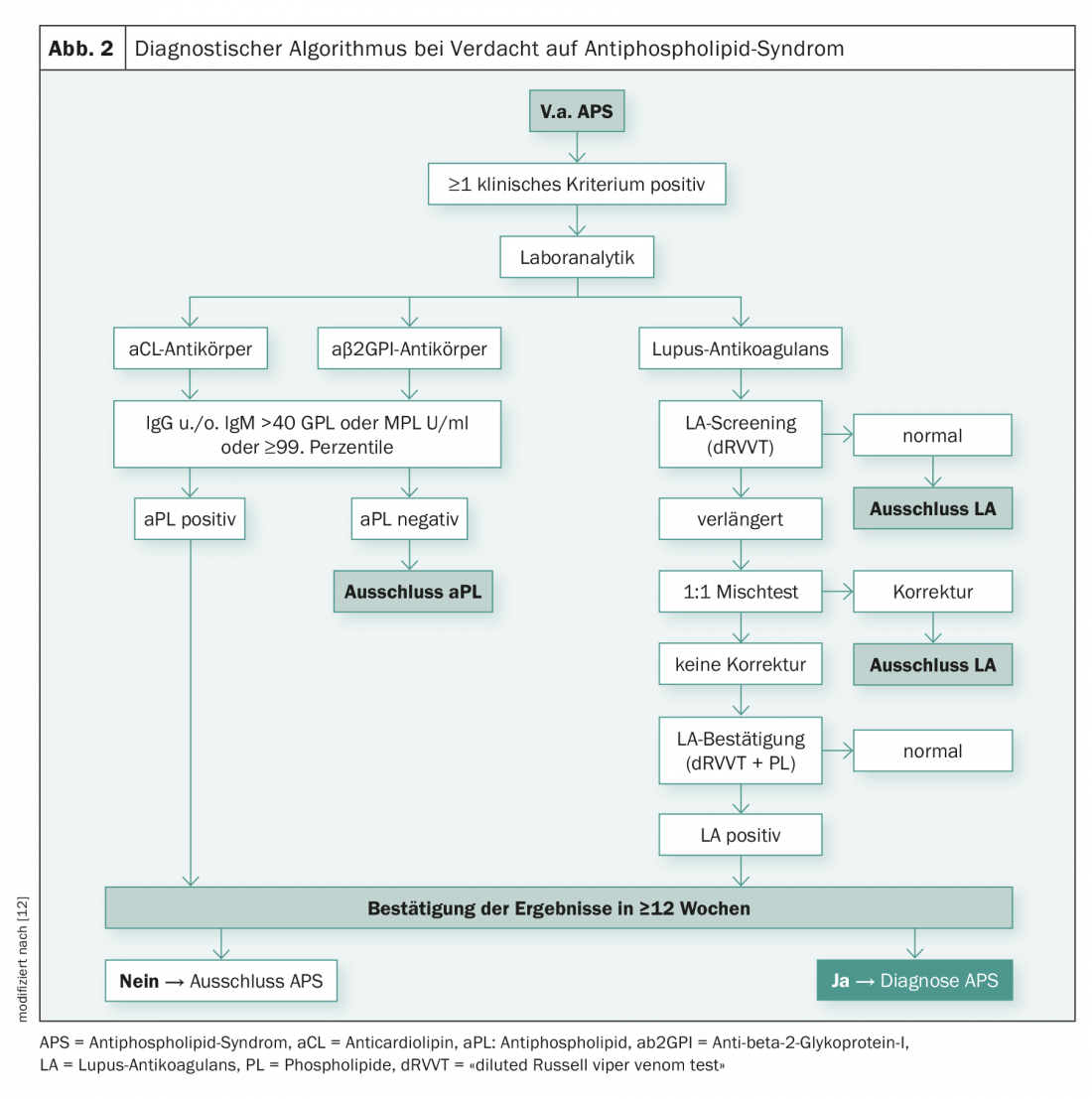
Test di coagulazione – Lupus anticoagulante: La Società Internazionale di Trombosi ed Emostasi (ISTH) definisce una strategia a tre fasi in due diverse procedure di test LA ciascuna [7]:
- Test di screening: prolungamento del tempo di coagulazione dipendente dai fosfolipidi (>99° percentile);
- Test misto: Conferma di un inibitore di ad azione immediata ed esclusione di una carenza di fattori della coagulazione;
- Test di conferma: Conferma che l’inibitore è fosfolipide-dipendente. La terza fase è necessaria per garantire che l’inibitore non sia diretto contro uno specifico fattore di coagulazione.
Test immunologici – aCL/aβ2GPI-ELISA: oltre ai test di coagulazione, gli anticorpi aPL (aCL-IgG e -IgM, nonché aβ2GPI-IgG e -IgM) vengono determinati immunologicamente mediante ELISA. In considerazione delle proprietà variabili dell’antigene e della mancanza di materiali di riferimento, la standardizzazione dei sistemi di test è molto difficile. I titoli anticorpali IgG e IgM sono espressi in unità GPL e MPL standardizzate a livello internazionale. I risultati positivi sono definiti come titoli ≥40 unità GPL/MPL (U/ml) o ≥99° percentile del rispettivo metodo [8].
Sebbene i test anti-β2GPI abbiano una maggiore specificità per quanto riguarda la trombogenicità rispetto all’aCL, non sono ancora sufficientemente standardizzati. I metodi di test che utilizzano il dominio ricombinante 1 della β2GPI, cioè l’epitopo per il legame con l’aPL, come antigene sono considerati promettenti [9,10]. Un lavoro recente ha dimostrato che gli anticorpi diretti contro questo epitopo sono più fortemente associati ai sintomi clinici. Tuttavia, sono necessari ulteriori sforzi per ottenere una migliore standardizzazione dei diversi metodi di test e una migliore caratterizzazione degli aPL clinicamente rilevanti.
Clinica
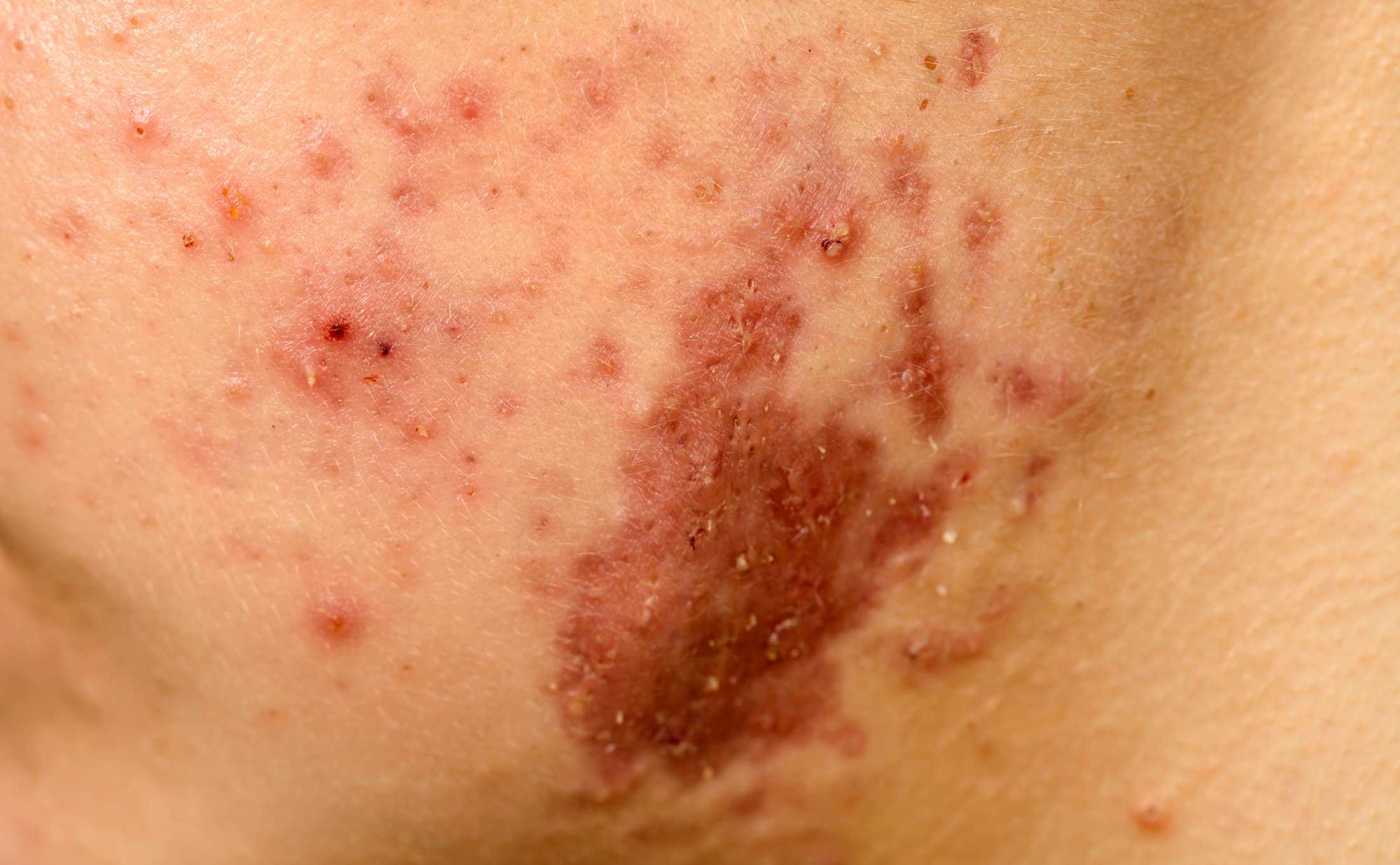
L’APS è una malattia multiorgano con manifestazioni multiple, classificata come trombofilia acquisita. Il quadro clinico è caratterizzato da trombosi arteriose e/o venose ricorrenti di vasi piccoli o grandi con localizzazione tipica o atipica, nonché da complicazioni vascolari della gravidanza. Inoltre, sono state descritte altre manifestazioni cliniche, come disfunzioni neurologiche (epilessia, demenza), sintomi dermatologici (livedo reticularis/racemosa, necrosi acrale), malattie cardiache valvolari (endocardite trombotica non batterica) e infarto miocardico, malattie renali (trombosi dell’arteria renale/vena renale, microangiopatia trombotica), malattie oculari (amaurosi fugace, occlusioni dei vasi retinici e/o coroideali) e trombocitopenie. In questi quadri clinici non è sempre chiaro se gli aPL rappresentino un epifenomeno o siano patogeneticamente coinvolti.
Indicazione: lo screening aPL deve essere avviato se esiste un’alta probabilità di presenza di APS in base a un criterio clinico (tab. 3) . Lo screening aPL non mirato in persone asintomatiche non è raccomandato, data la bassa specificità dei sistemi di test.
La diagnosi di APS si basa sul rilevamento ripetuto di LA, aCL o aβ2GPI a intervalli di 12 settimane, come già detto. Il profilo aPL (isotipo e numero di test positivi) può essere utilizzato anche per valutare il rischio e l’affidabilità dei risultati [11]. È stato dimostrato che il 98% dei pazienti triplo positivi, l’84% di quelli doppio positivi e il 40% di quelli singolo positivo erano ancora positivi dopo dodici settimane. Questi dati sottolineano che il profilo triplo positivo è un risultato di laboratorio solido. Inoltre, un profilo triplo positivo corrisponde a una classificazione per APS ad alto rischio, mentre un profilo doppio o singolo positivo corrisponde a un rischio moderato o basso di APS, rispettivamente.
Terapia
La strategia di trattamento quando viene rilevato un aPL dipende principalmente dal fatto che i pazienti siano asintomatici con solo risultati di laboratorio positivi o sintomatici con complicazioni manifeste. Per i pazienti asintomatici, vale la stessa raccomandazione di tromboprofilassi che si applica agli altri pazienti con trombofilia. Nel caso di APS manifesta con sintomi corrispondenti, tuttavia, bisogna distinguere se è presente la forma primaria o secondaria. Nel caso di APS primaria senza evidenza di un’altra malattia di base, la terapia è finalizzata esclusivamente a prevenire ulteriori complicazioni della trombosi. Nel caso di APS secondaria, il primo passo è trattare la malattia di base o minimizzare i fattori di rischio tipici per le complicanze cardiovascolari, parallelamente al trattamento della sindrome trombotica.
Anticoagulazione (ASA, VKA, NOAK): nei pazienti APS con trombosi venosa, si raccomanda una terapia iniziale con eparina non frazionata (UFH) o a basso peso molecolare (NMH), seguita da una terapia a lungo termine con antagonisti della vitamina K (VKA). A causa dell’elevato rischio di trombosi ricorrente (che dipende, tra l’altro, dall’appartenenza del paziente al gruppo ad alto rischio con risultati aPL tripli positivi) dopo l’interruzione dell’anticoagulazione orale, l’anticoagulazione orale a lungo termine con INR 2-3 target è la terapia di scelta nella maggior parte dei casi, almeno finché i risultati di laboratorio rimangono positivi [12]. Questo riduce in modo significativo le recidive tromboemboliche. Tuttavia, in alcuni casi si rivela insufficiente. In questi casi, può essere presa in considerazione una terapia a lungo termine con NMH o l’innalzamento dell’INR target a 3-4.
I pazienti con trombosi arteriosa o con APS tripla positiva che sono ad alto rischio di eventi tromboembolici traggono maggiore beneficio dall’anticoagulazione sistemica che dagli agenti antiaggreganti. In questi pazienti è indicata l’anticoagulazione a vita.
L’uso dei nuovi anticoagulanti orali diretti (NOAK) è controverso [12]. I NOAK non richiedono controlli di laboratorio e sono risultati efficaci e sicuri almeno quanto i VKA o i NMH nel trattamento o nella prevenzione del tromboembolismo venoso e arterioso. Tuttavia, studi prospettici e retrospettivi hanno dimostrato che i NOAK sono meno efficaci del warfarin nei pazienti con APS ad alto rischio [13,14]. L’attuale base di prove per l’uso e l’efficacia dei NOAK nei pazienti con APS è ancora insufficiente e sono necessari ulteriori studi prospettici per valutare il loro valore nell’APS.
L’anticoagulazione nelle donne con complicazioni ricorrenti della gravidanza dovute all’APS è finalizzata a preservare il feto o ad aumentare il tasso di nati vivi. Per il trattamento delle donne in gravidanza, si raccomanda una combinazione di NMH a dosaggio sub-terapeutico (1×100 E/kgKG/giorno) e aspirina (1×100 mg/giorno). L’anticoagulazione deve essere iniziata il prima possibile dopo la conferma della gravidanza e deve essere continuata fino a sei settimane dopo il parto.
Immunosoppressione: il trattamento con immunosoppressori in aggiunta all’anticoagulazione è indicato principalmente in presenza di APS secondaria, trombocitopenia o sindrome antifosfolipidica catastrofica (CAPS). Si sta discutendo anche l’uso di immunosoppressori nell’APS per la profilassi di ulteriori eventi tromboembolici, in quanto l’immunosoppressione ha un’influenza favorevole sugli alti titoli di aPL e potrebbe quindi risultare in una minore incidenza di occlusione vascolare. Tuttavia, non sono disponibili raccomandazioni concrete sul tipo e la durata dell’immunosoppressione. Questi derivano dall’esperienza nel trattamento della trombocitopenia autoimmune o degli inibitori dei fattori della coagulazione (ad esempio, terapia singola o combinata con prednisone/ciclofosfamide/rituximab).
Messaggi da portare a casa
- La sindrome antifosfolipidica (APS) è una malattia autoimmune sistemica con un evento patogenetico primario: la formazione di autoanticorpi contro i complessi fosfolipidi/proteine attivi nei coaguli.
- Nonostante il fenomeno paradossale del prolungamento dei test di coagulazione globale in vitro, il quadro clinico del paziente con APS non è caratterizzato da emorragie, ma da manifestazioni trombotiche.
- La diagnosi di APS richiede risultati di laboratorio (lupus anticoagulante e/o anticorpi antifosfolipidi medio-alti positivi) combinati con una trombosi clinica nel micro o macrocircolo.
- L’APS è considerata una trombofilia acquisita e necessita di una tromboprofilassi farmacologica variabile come terapia, adattata per intensità e durata, a seconda del coinvolgimento degli organi e dell’entità delle manifestazioni trombotiche. Le eparine o gli antagonisti della vitamina K restano gli anticoagulanti di prima scelta.
Letteratura:
- Cervera R, Espinosa, Khamashta MA: Sindrome antifosfolipidica nelle malattie autoimmuni sistemiche, 2a edizione. Elsevier, Amsterdam, 2016.
- Giannakopoulos B, Krilis SA: La patogenesi della sindrome antifosfolipidica. N Engl J Med 2013; 368: 1033-1044.
- Asherson RA, et al: Sindrome antifosfolipidica catastrofica: dichiarazione di consenso internazionale sui criteri di classificazione e sulle linee guida di trattamento. Lupus 2003; 12: 530-534.
- Arnout J, Vermylen J: Stato attuale e implicazioni degli anticorpi autoimmuni antifosfolipidi in relazione alla malattia trombotica; J Thromb Heamost 2003; 1: 931-942.
- Wilson WA, et al: Dichiarazione di consenso internazionale sui criteri di classificazione preliminare per la sindrome antifosfolipidica definita: relazione di un workshop internazionale. Arthritis Rheum 1999; 42: 1309-1311.
- Miyakis S, et al: Dichiarazione di consenso internazionale su un aggiornamento dei criteri di classificazione per la sindrome antifosfolipidica definita (APS). J Thromb Haemost 2006; 4: 295-306.
- Pengo V, et al.: Aggiornamento delle linee guida per il rilevamento del lupus anticoagulante. Sottocomitato sul lupus anticoagulante/anticorpo antifosfolipide del Comitato scientifico e di standardizzazione della Società internazionale sulla trombosi e l’emostasi, J Thromb Haemost 2009; 7: 1737-1740.
- Devreese KMJ, et al:. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibodies, Criteri di laboratorio per la sindrome antifosfolipidica: comunicazione del SSC dell’ISTH, J Thromb Haemost 2018; 16: 809-813.
- De Craemer AS, Musial J, Devreese KM: Ruolo degli anticorpi anti-dominio 1-beta2 glicoproteina I nella diagnosi e nella stratificazione del rischio della sindrome antifosfolipidica. J Thromb Haemost 2016; 14: 1779-1787.
- Pengo V, et al: Sindrome antifosfolipidica: gli anticorpi al dominio 1 della beta2-glicoproteina 1 classificano correttamente i pazienti a rischio. J Thromb Haemost 2015; 13:782-7.
- Pengo V, et al.: La conferma della positività iniziale degli anticorpi antifosfolipidi dipende dal profilo degli anticorpi antifosfolipidi. J Thromb Haemost 2013; 11: 1-5.
- Garcia D, Erkan D: Diagnosi e gestione della sindrome antifosfolipidica. N Engl J Med 2018; 378: 2010-2021.
- Martinelli I, et al: Trombosi ricorrente nei pazienti con anticorpi antifosfolipidi trattati con antagonisti della vitamina K o rivaroxaban. Haematologica 2018;103(7): 315-317.
- Pengo V, et al: Rivaroxaban vs warfarin nei pazienti ad alto rischio con sindrome antifosfolipidica. Sangue 2018; 132(13): 1365-1371.
- Tsakiris D, Bachofner A: Coagulazione intravascolare disseminata nel paziente con tumore. Info@Oncology 2017; 7:19-21.
InFo ONcOLOGIA & EMATOLOGIA 2019; 7(1): 22-25.