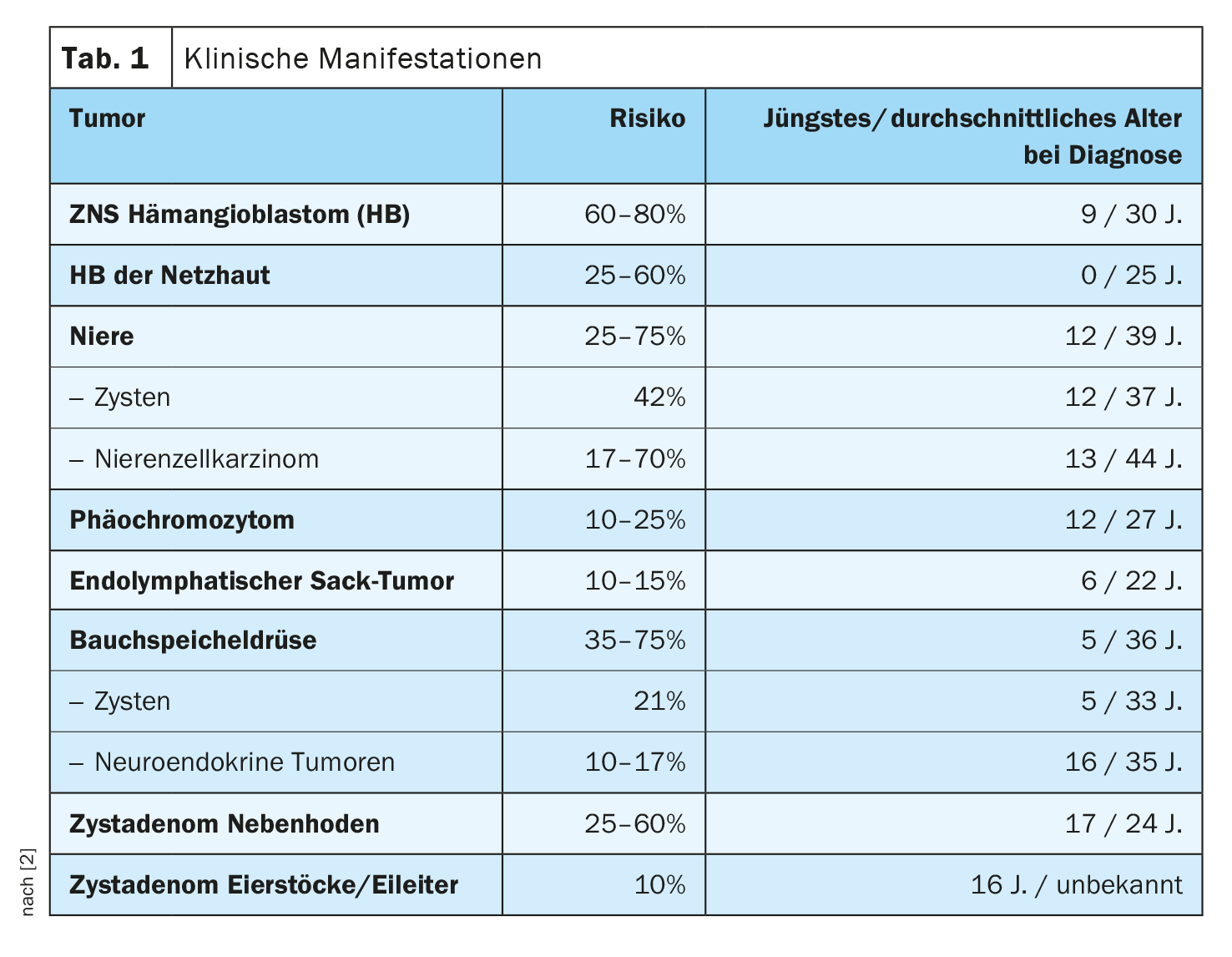
La sindrome di Von Hippel-Lindau è dovuta a mutazioni nel gene VHL ed è caratterizzata da emangioblastomi del cervello, del midollo spinale e della retina dell’occhio. Inoltre, i soggetti affetti presentano un rischio maggiore di carcinoma a cellule renali e cisti renali, feocromocitomi, cisti e tumori neuroendocrini del pancreas, tumori del sacco endolinfatico e cisti dell’epididimo, nonché delle ovaie e delle tube di Falloppio.
I sintomi dipendono dalle dimensioni e dalla posizione dei tumori [1]. I bambini possono soffrire di mal di testa e provare vertigini o debolezza. Inoltre, possono verificarsi disturbi visivi, con possibile perdita della vista nei tumori della retina in crescita, e pressione alta. Potrebbe esserci una perdita di coordinazione. Circa il 10% dei bambini affetti presenta un tumore dell’orecchio interno, che può compromettere l’udito. Senza un trattamento, i pazienti possono diventare ciechi, subire danni cerebrali o morire. I decessi sono di solito il risultato di complicazioni di angiomi cerebrali o di cancro ai reni.

La diagnosi precoce è di grande importanza. Fenotipicamente, si distingue tra diversi tipi di sindrome di Von Hippel-Lindau [1]: Il tipo I è caratterizzato dalla presenza di emangiomi nella retina e/o nel sistema nervoso centrale, carcinomi a cellule renali e/o tumori neuroendocrini. Tuttavia, il rischio di feocromocitoma è molto basso. Il tipo I è associato a mutazioni nonsense o a delezioni di segmenti genici più grandi. Al contrario, il tipo II è spesso associato a mutazioni missense e il rischio di sviluppare feocromocitomi è molto alto.
Criteri diagnostici
La diagnosi di sindrome di Von Hippel-Lindau è considerata confermata se viene rilevata una mutazione nel gene VHL e/o [2]:
Senza sindrome di Von Hippel-Lindau nella famiglia in presenza di almeno 2 dei seguenti risultati:
≥2 emangioblastomi della retina, del midollo spinale o del cervello, oppure un singolo emangioblastoma insieme a una manifestazione nell’addome (ad esempio, cisti multiple dei reni o del pancreas).
– Carcinoma a cellule renali
– Feocromocitoma
– ELST, cistoadenoma delle ovaie o delle tube di Falloppio/epididimo o tumori neuroendocrini del pancreas
Con la sindrome di Von Hippel-Lindau nella famiglia in presenza di almeno. 1 dei seguenti risultati:
– Emangioblastoma della retina
– Emangioblastoma del midollo spinale o del cervelletto
– Feocromocitoma
– Carcinoma a cellule renali
– Cisti multiple dei reni o del pancreas
Letteratura:
- Institut für Klinische Genetik, Universitätsklinikum Carl Gustav Carus Dresden, www.uniklinikum-dresden.de, (ultimo accesso 17.01.2023)
- Medizinische Hochschule Hannover, www.krebs-praedisposition.de/fuer-patienten-und-familien/von-hippel-lindau-syndrom/#diagnose, (ultimo accesso 17.01.2023)
- Baumgartner-Parzer S: J Clin Endocrinol Stoffw 2020; 13: 37-40.
HAUSARZT PRAXIS 2023; 18(1): 47
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(1): 32