
Le cellule T dotate di specificità tumorale attraverso l’espressione di un recettore antigenico chimerico (CAR) stanno diventando sempre più importanti. Sono già sempre più utilizzate nella terapia cellulare adottiva nella lotta contro il cancro. Il vantaggio principale del trasferimento di un CAR rispetto al trasferimento di un normale recettore delle cellule T (TCR) è che un CAR può riconoscere il tumore indipendentemente dall’MHC.
Le cellule T dotate di specificità tumorale attraverso l’espressione di un recettore antigenico chimerico (CAR) stanno diventando sempre più importanti. Sono già sempre più utilizzate nella terapia cellulare adottiva nella lotta contro il cancro. Il concetto di CAR è stato originariamente sviluppato alla fine degli anni ’80 da Zelig Eshhar (Istituto Weizmann di Scienze, Rehovot, Israele) [1,2]. La maggior parte dei CAR consiste in un costrutto scFv legato all’antigene tumorale e derivato da un anticorpo. (frammento variabile a catena singola, che è una proteina di fusione prodotta artificialmente, composta da una parte variabile di una catena leggera e pesante di un’immunoglobulina) e dalla parte intracellulare della catena CD3ζ, il in do diesis è legato a uno o più domini costimolatori [3]. Questa struttura a blocchi consente l’attivazione delle cellule T antigene-specifiche in risposta al riconoscimento specifico degli antigeni sulla superficie delle cellule maligne, avviato dal legame di scFv e dalla successiva segnalazione attraverso la catena CD3ζ e il dominio costimolatorio [3]. La costimolazione avviene solitamente tramite il CD28 (superfamiglia delle immunoglobuline) o tramite il 4-1BB (superfamiglia del recettore del TNF) [3]. Tuttavia, esistono anche molti altri formati. Il vantaggio principale del trasferimento di un CAR rispetto al trasferimento di un normale recettore delle cellule T (TCR) è che un CAR può riconoscere il tumore indipendentemente dall’MHC.
Questa tecnologia è stata finora utilizzata per sviluppare i CAR che mirano a vari antigeni della superficie cellulare dei tumori solidi o ematologici. Le cellule CAR-T, specifiche per gli antigeni bersaglio come il CD19 sulle cellule B o l’antigene di maturazione delle cellule B (BCMA) sulle plasmacellule, hanno portato a regressioni cliniche impressionanti nelle leucemie, nei linfomi o nei mielomi in diversi studi clinici [4–6]. Risultati come questi hanno portato, tra l’altro all’approvazione di tisagenlecleucel per il trattamento della leucemia linfoblastica acuta a cellule B (ALL), axicabtagene-ciloleucel per il trattamento del linfoma non-Hodgkin aggressivo a cellule B, Brexucabtagene-Autoleucel per il trattamento del linfoma a cellule del mantello, Lisocabtagene-Maraleucel per il trattamento del linfoma a grandi cellule B e Idecabtagene-Vicleucel e Ciltacabtagene-Autoleucel per il trattamento del mieloma multiplo. Food and Drug Administration degli Stati Uniti (FDA) e il Agenzia Europea dei Medicinali (EMA) [3].
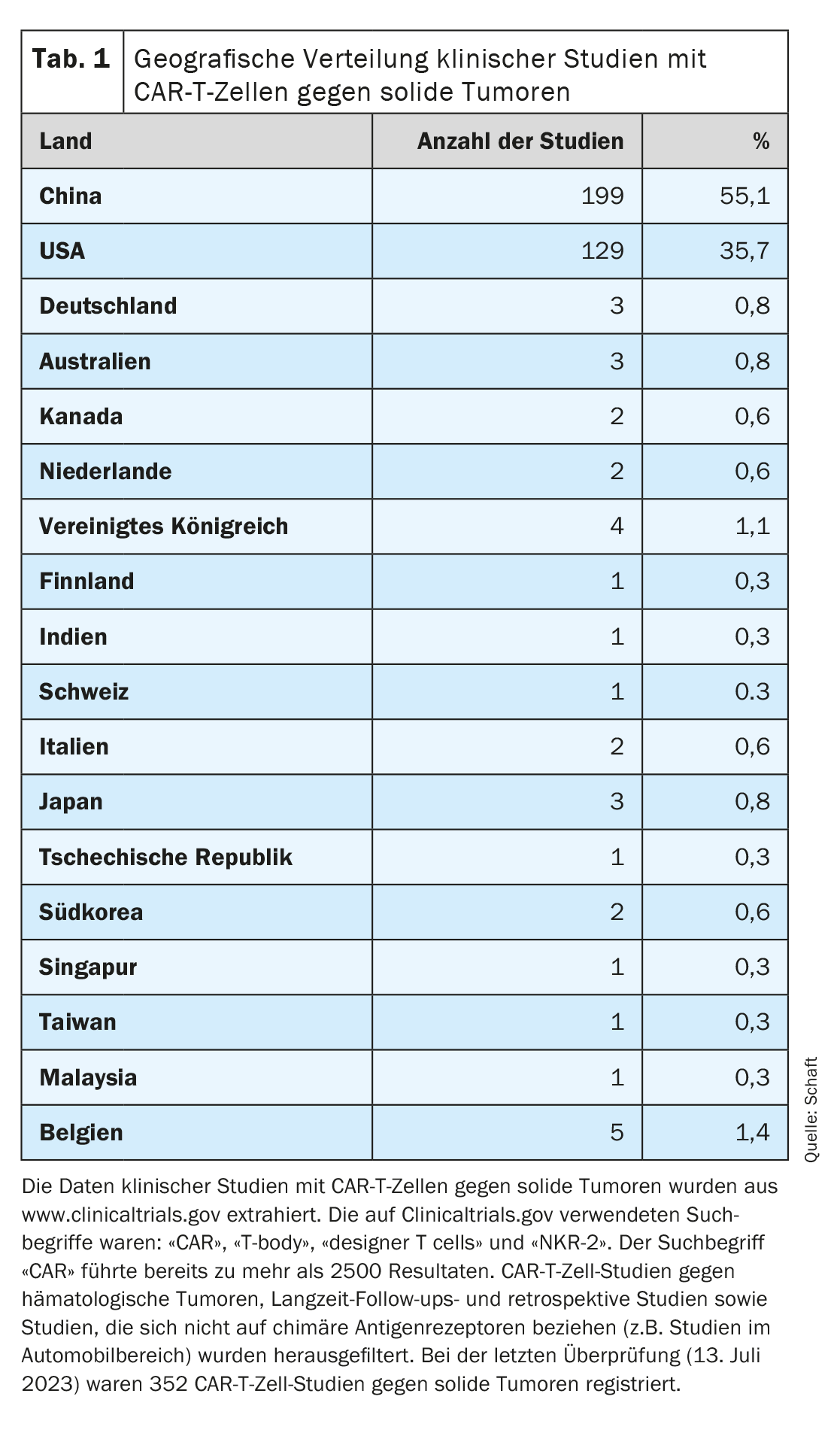
Poiché la maggior parte degli studi clinici si concentra sull’eliminazione dei tumori ematologici, lo sviluppo delle cellule CAR-T contro i tumori solidi è in ritardo (panoramica dettagliata in [7–11]). Osservando la distribuzione geografica delle sperimentazioni cliniche con cellule CAR-T contro i tumori solidi registrate su Clinicaltrials.gov (n=352; ultima valutazione il 13 luglio 2023), è chiaro che la maggior parte di queste sperimentazioni è condotta in Cina (n=199; 55,1%). Gli Stati Uniti sono al secondo posto (n=129; 35,7%). Solo pochissimi studi sono stati condotti in Europa (Germania n=3, Svizzera n=1), Australia e nel resto dell’Asia (combinato n=33; 9,2%) (Tabella 1).
Formati CAR
Dalla pubblicazione del primo concetto di CAR da parte di Zelig Eshhar nel 1989 [1,2], i CAR sono stati continuamente sviluppati. Questo ha portato a diverse generazioni di CAR basate sulla struttura di base del concetto originale di CAR. Il CAR classico contiene sempre un anticorpo scFv in grado di legare l’antigene tumorale. Nei CAR di prima generazione (Fig. 1), lo scFv è collegato al dominio di segnalazione intracellulare di FcεRIγ o CD3ζ tramite un linker flessibile e un dominio transmembrana [11,12].
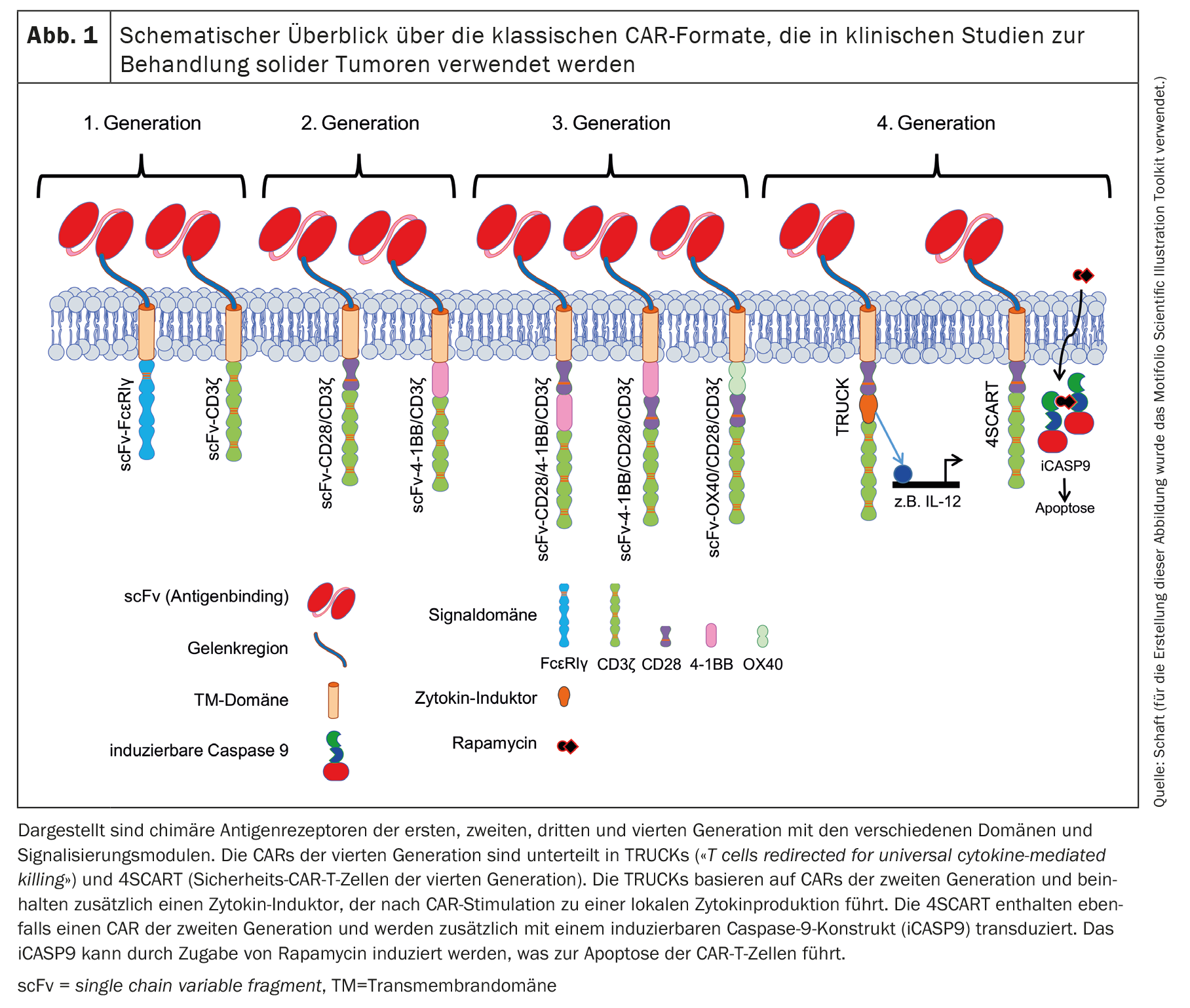
La maggior parte delle sperimentazioni cliniche registrate con cellule CAR-T contro i tumori solidi utilizza un CAR [11,12] di seconda generazione, che contiene anche un dominio costimolatorio (Fig. 1). La costimolazione è solitamente effettuata da CD28 o 4-1BB [3]. La co-stimolazione del CD28 supporta fisiologicamente la produzione di IL-2, -6, -10 e altre interleuchine, nonché la progressione del ciclo cellulare, la sopravvivenza, la differenziazione e la funzione citolitica delle cellule T [13]. In molti studi in cui sono stati utilizzati CAR con un dominio di segnalazione CD28, sono stati osservati effetti antitumorali efficaci e rapidi. Tuttavia, questi sono stati solo di breve durata e sono stati associati a una sopravvivenza limitata in vivo rispetto ai CAR con un dominio di segnalazione 4-1BB, ad esempio [14]. La segnalazione fisiologica di 4-1BB nelle cellule T aumenta la progressione del ciclo cellulare e la proliferazione, la secrezione di citochine, il potenziale citolitico delle cellule T e inibisce la cancellazione clonale e lamorte cellulare indotta dall’attivazione (AICD) [15,16]. I CAR contenenti 4-1BB come dominio di segnalazione non solo hanno consentito un’attivazione cellulare più robusta, una maggiore persistenza in vivo, ma hanno anche promosso la differenziazione delle cellule CAR-T verso lecellule della memoria centrale [4,14,17–24].
I CAR di terza generazione [11,12] contengono combinazioni di domini costimolatori: CD28/4-1BB, 4-1BB/CD28 o OX40/CD28 (Fig. 1) [25,26]. I CAR di quarta generazione sono fondamentalmente CAR di seconda generazione con caratteristiche aggiuntive. Le TRUCK (cellule T reindirizzate per l’uccisione universale mediata da citochine) sono modificate in modo tale da produrre citochine in modo localizzato molto limitato [27]. Gli effetti indotti dipendono dal tipo di citochine rilasciate: L’IL-12, ad esempio, può attivare una risposta immunitaria innata contro il tumore [28], provoca una ridotta suscettibilità agli effetti inibitori da parte delle cellule T regolatorie (Tregs) [29] e aumenta la secrezione di citochine e la proliferazione delle cellule T [30,31]. L’IL-15, invece, aumenta l’attività antitumorale delle cellule CAR-T [32].
Un’altra variante della quarta generazione è la 4SCART (cellule CAR-T di sicurezza). Queste cellule T sono trasdotte contemporaneamente con un CAR e una caspasi 9 inducibile (iCASP9) come precauzione di sicurezza contro gli eventi avversi. L’iCASP9 può essere indotto dall’aggiunta di rapamicina, che porta all’apoptosi delle cellule CAR-T.
Tecnologie di trasferimento
Un requisito essenziale per la produzione di cellule T CAR è quello di trovare un metodo adatto per trasferire il CAR nelle cellule T. A questo scopo, si possono utilizzare diversi metodi esistenti. La maggior parte delle sperimentazioni cliniche utilizza un metodo di trasferimento virale (retrovirale o lentivirale) per introdurre stabilmente il CAR nelle cellule T. Durante questo processo, un gene che codifica CAR viene trasportato dal virus nella cellula T, dove viene integrato in modo stabile nel DNA genomico. La progenie di queste cellule trasdotte porta tutte il gene CAR e può esprimere il recettore sulla sua superficie cellulare. Gli svantaggi della trasduzione virale sono l’integrazione casuale nel genoma della cellula ospite, che può portare alla distruzione o all’attivazione di alcuni geni (cioè la mutagenesi inserzionale), nonché l’introduzione di materiale/geni virali. Questo metodo può portare a gravi effetti collaterali nei pazienti trattati con cellule CAR-T. Lamers et al. hanno descritto, ad esempio, lo sviluppo di risposte immunitarie al transgene codificante il recettore e al vettore retrovirale [33].
Alcune sperimentazioni cliniche utilizzano un sistema di consegna del gene non virale o un metodo di trasferimento che integra il gene CAR in un sito specifico (ad esempio, il sistema di trasposoni Sleeping Beauty [34–37], il sistema di trasposoni PiggyBac [36,37], CRISPR-Cas9 [38]). La trasfezione del DNA o dell’RNA sono ulteriori sistemi di trasferimento [39], che tuttavia non portano all’integrazione della sequenza codificante CAR nel genoma della cellula ospite. La conseguente espressione transitoria del CAR presenta alcuni vantaggi.
Cellule CAR-T contro i tumori solidi – selezione dell’antigene e precauzioni di sicurezza
Come descritto sopra, l’uso clinico delle cellule CAR-T nel trattamento dei tumori solidi è in ritardo rispetto al successo delle cellule CAR-T nel trattamento dei tumori ematologici. Una delle ragioni è che il CD19 e il BCMA sono antigeni bersaglio espressi in modo specifico dalle cellule B o dalle plasmacellule e la loro completa eliminazione è relativamente innocua. Altri antigeni, soprattutto nei tumori solidi, sono spesso espressi anche nei tessuti sani, il che rende difficile selezionare un antigene target adatto.
Selezione dell’antigene
Gli antigeni target ideali dei tumori solidi combinano tre proprietà essenziali:
- Espressione uniforme sulla superficie delle cellule maligne, che riduce il rischio di varianti di fuga antigene-negative.
- La mancanza di espressione sulle cellule non maligne (cioè l’espressione esclusiva sulle cellule tumorali), previene il rischio di effetti collaterali gravi e potenzialmente fatali derivanti dall’attività on-target/off-tumour delle cellule CAR-T [40,41].
- Un ruolo cruciale come driver oncogenico nelle cellule tumorali che impedisce la downregulation dell’antigene a causa di un vantaggio selettivo di sopravvivenza delle cellule maligne.
- Inoltre, la co-espressione dell’antigene sulle cellule vicine all’interno del microambiente tumorale (ad esempio, sui vasi associati al tumore, sui fibroblasti e sui macrofagi) è un’altra caratteristica positiva di un antigene bersaglio ideale, in quanto la struttura di approvvigionamento del tumore può essere attaccata dalla terapia antigene-specifica.
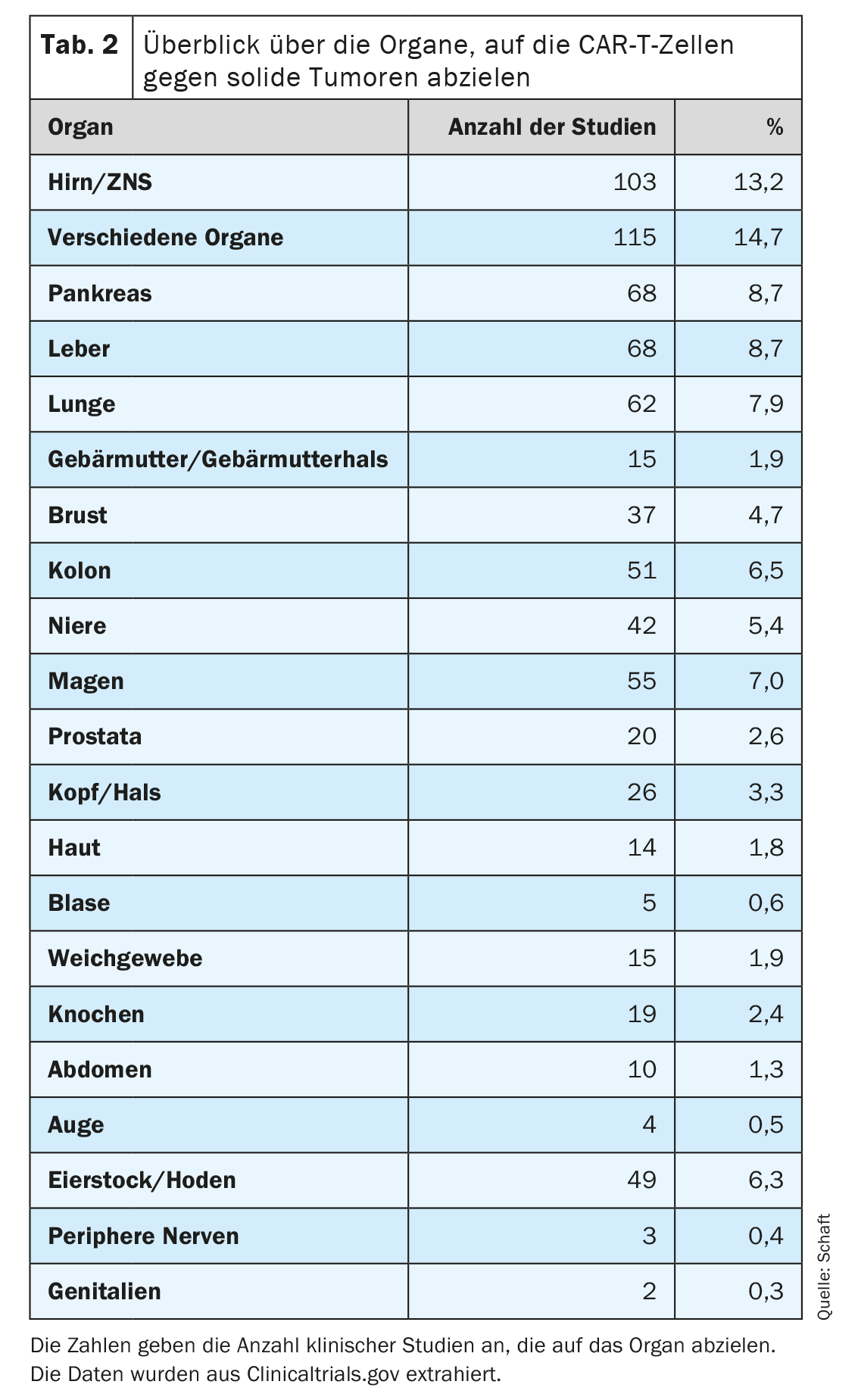
Il secondo punto, in particolare, rappresenta il problema maggiore per lo sviluppo delle cellule CAR-T contro i tumori solidi, poiché la maggior parte degli antigeni espressi nei tumori solidi sono espressi anche in importanti tessuti sani. Questo può portare a una reazione indesiderata on-target/off-tumour e agli effetti collaterali associati. Tuttavia, molti tipi diversi di tumori solidi (81 entità tumorali in totale) in un totale di 20 organi sono stati presi di mira con cellule CAR-T specifiche per 63 antigeni diversi (Tabella 2). In molti studi clinici, vengono studiati in particolare i tumori del cervello/CNS, del fegato, del pancreas e del polmone (rispettivamente n=103, 68, 68 e 62); Scheda. 2). Ciò potrebbe essere dovuto all’elevata necessità medica e/o alla mancanza di terapie alternative efficaci per i tumori negli organi corrispondenti.
Precauzioni di sicurezza
Se un antigene bersaglio viene riconosciuto dalle cellule CAR-T trasferite su un tessuto sano, possono verificarsi effetti collaterali indesiderati e gravi. Sono state sviluppate diverse strategie per spegnere le cellule CAR-T il più rapidamente possibile in caso di tossicità nel paziente. La rapamicina, una molecola in grado di indurre la dimerizzazione dei costrutti, può essere utilizzata, ad esempio, per attivare una caspasi 9 inducibile. Nella 4SCART, questi costrutti inducibili vengono trasferiti nelle cellule T contemporaneamente al CAR come un cosiddetto interruttore suicida (Fig. 1). Dopo la dimerizzazione indotta dalla rapamicina, la caspasi 9 induce l’apoptosi delle cellule T CAR. In questo modo si elimina anche l’attività indesiderata/imprevista delle cellule T contro i tessuti sani(effetti on-target/off-tumour) [42,43]. Altri possibili switch, come la strategia della timidina chinasi/ganciclovir del virus herpes simplex (HSV-tk/GCV) [44,45] sono già in uso [11].
Una misura di sicurezza speciale per aggirare l’autoimmunità prolungata indotta da una reazione on-target/off-tumour del CAR è la trasfezione del CAR mediante elettroporazione di mRNA [11]. Abbiamo già dimostrato in diverse pubblicazioni che la trasfezione transitoria delle cellule T con i CAR utilizzando l’elettroporazione dell’mRNA può essere uno strumento efficace e sicuro nell’immunoterapia del cancro [46-50]. Il processo di elettroporazione si basa su complessi meccanismi fisico-chimici che portano alla perforazione della membrana plasmatica attraverso l’applicazione di campi elettrici e consentono il successivo ingresso dell’mRNA nel citosol [51]. L’uso di cellule CAR-T trasfettate con RNA offre il vantaggio che l’espressione del recettore è limitata nel tempo, in modo da limitare anche la potenziale tossicità off-target e on-target/off-tumour. La strategia di trasferimento del CAR-RNA è particolarmente interessante negli studi clinici di fase 0/1 che studiano nuovi antigeni tumorali per la terapia con cellule T CAR con un profilo di sicurezza clinica sconosciuto.
Cellule CAR-T clinicamente testate contro il melanoma uveale
Sorprendentemente, solo quattro studi clinici sulle cellule CAR T contro i tumori solidi si concentrano sull’occhio (Tabella 2) . Di questi, due studi sono diretti contro il retinoblastoma e due studi contro il melanoma uveale. Il melanoma uveale è il tipo più comune di tumore dell’occhio e presenta metastasi fino al 50% dei pazienti. Le metastasi si verificano prevalentemente nel fegato e sono associate a una scarsa sopravvivenza mediana di circa 12 mesi. Nonostante gli enormi progressi nel trattamento del melanoma cutaneo metastatizzato con il blocco del checkpoint immunitario (ICB), non è efficace nel melanoma uveale. Solo l’ingaggiatore bispecifico di cellule T Tebentafusp, approvato di recente (uno scFv specifico per CD3 collegato a un TCR solubile che riconosce un peptide gp100 presentato da HLA-A2), può attenuare la progressione e prolungare la sopravvivenza globale in un sottogruppo di pazienti con melanoma uveale metastatico. Gli effetti positivi osservati di Tebentafusp sono di breve durata, con un tempo di sopravvivenza globale mediano di 22 mesi e un tasso di sopravvivenza a tre anni del 24%. Inoltre, solo il 50% dei pazienti metastatizzati è idoneo a questa opzione terapeutica a causa della restrizione HLA-A2. Esiste quindi anche un’elevata necessità medica di approcci terapeutici alternativi per questa entità tumorale.
Due studi clinici con cellule CAR-T elencati nella National Library of Medicine degli Stati Uniti (www.clinicaltrials.gov) stanno attualmente reclutando pazienti con melanoma uveale (NCT03893019 contro il cluster di differenziazione 20 [CD20] e NCT03635632 contro il disialoganglioside GD2 [GD2]). Entrambi gli studi utilizzano cellule CAR-T che mirano ad antigeni non specifici del melanoma.
Il primo studio clinico (fase 1; NCT03893019), che utilizza cellule CAR-T specifiche per CD20 di seconda generazione (Fig. 2A), è sponsorizzato da Miltenyi Biomedicine GmbH (Principal Investigator [PI]: Peter Borchmann; Ospedale Universitario di Colonia) e sta reclutando principalmente pazienti con melanoma cutaneo. Vengono trattati anche alcuni pazienti con melanoma uveale. Il CD20 è un antigene bersaglio che viene espresso sulle cellule B normali ed è tipicamente utilizzato come antigene bersaglio nel linfoma non-Hodgkin a cellule B [52]. Tuttavia, è espresso anche in un piccolo sottogruppo di cellule di melanoma [53,54]. Tuttavia, il trattamento mirato di un antigene che è espresso solo su un piccolo sottogruppo di cellule tumorali potrebbe far sì che il tumore sfugga facilmente alla terapia con cellule CAR-T. Purtroppo, lo stato attuale di questo studio clinico non è noto.
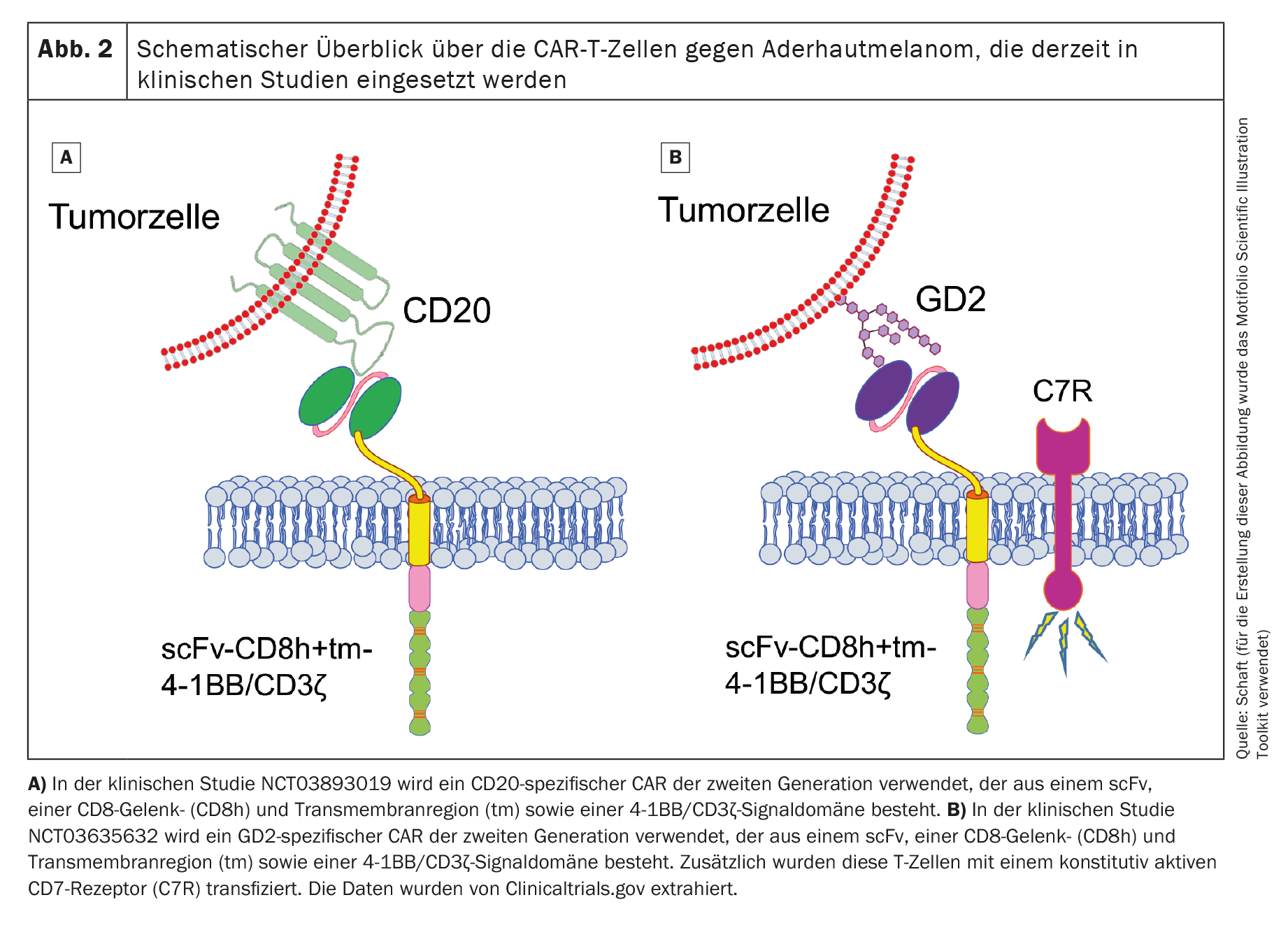
Il secondo studio clinico (anch’esso di fase 1; NCT03635632) utilizza cellule CAR-T specifiche per GD2 (Fig. 2B) e sta reclutando pazienti con neuroblastoma, sarcoma, melanoma uveale, cancro al seno o altri tumori che esprimono GD2. Questo studio, sponsorizzato dal Baylor College of Medicine (PI: Bilal Omer; Baylor College of Medicine), è attualmente attivo, ma non sono stati reclutati pazienti.
Oltre al CAR di seconda generazione, i ricercatori hanno anche trasdotto un recettore IL-7 costitutivamente attivo nelle cellule T, per prolungare la sopravvivenza delle cellule CAR-T dopo il trasferimento adottivo. Il GD2 è espresso, anche se a livelli molto bassi, nel cervelletto e nei nervi periferici [55], il che rende il trattamento con cellule CAR-T GD2-specifiche molto rischioso se viene indotta una reazione on-target/off-tumour. Anche per questo studio clinico non sono ancora stati pubblicati dati.
In sintesi, esiste un grande potenziale per gli studi clinici con cellule CAR-T contro antigeni specifici del melanoma (uvea), dove il rischio di una reazione on-target/off-tumour è basso.
La ricerca di un migliore antigene tumorale nel melanoma uveale
La prevenzione o la riduzione di una possibile reazione on-target/off-tumour è, come già detto nella sezione “Cellule CAR-T contro i tumori solidi”, un requisito fondamentale nella ricerca di nuovi antigeni. A livello preclinico, l’attenzione si concentra attualmente su due antigeni espressi nei melanomi uveali: il recettore 2 del fattore di crescita epidermico umano (HER2) e il condroitin solfato proteoglicano 4 (CSPG4).
HER2
HER2 è un membro della famiglia ErbB delle tirosin-chinasi recettoriali (EGFR [ErbB-1], HER2 [/neu] [ErbB-2], Her 3 [ErbB-3] e Her 4 [ErbB-4]). Le mutazioni in HER2 portano alla sovraespressione, con conseguente attivazione costitutiva e divisione cellulare incontrollata. Questo vale soprattutto per il cancro al seno, ma anche per altri tipi di tumore, come il cancro ovarico o i gliomi [56–58].
Come già detto, l’uso delle cellule CAR-T è un’arma a doppio taglio, poiché l’efficacia di queste cellule può anche ritorcersi contro il paziente [59]. Non si può mai escludere che un tipo di cellula rara ma essenziale esprima l’antigene nel tessuto sano. I ricercatori del National Cancer Institute hanno riportato un caso che illustra il potenziale mortale della tossicità on-target/off-tumour dell’antigene HER2. Poco dopo l’infusione di cellule CAR-T HER2-specifiche, sono stati osservati sintomi clinici di sindrome da distress respiratorio acuto che hanno richiesto la respirazione artificiale in un paziente con cancro colorettale metastatico [60]. Purtroppo, il paziente è morto cinque giorni dopo l’insorgenza del distress respiratorio acuto [60]. La causa del decesso è stata probabilmente il risultato della tossicità on-target/off-tumour causata da bassi livelli di HER2 sulle cellule epiteliali dei polmoni. È sorprendente che il CAR sia stato basato sull’anticorpo monoclonale trastuzumab, approvato dalla FDA, che è stato utilizzato ampiamente senza causare una grave tossicità polmonare [61]. Questo sottolinea la necessità di una selezione molto accurata dell’antigene bersaglio per la terapia con cellule T CAR.
CSPG4
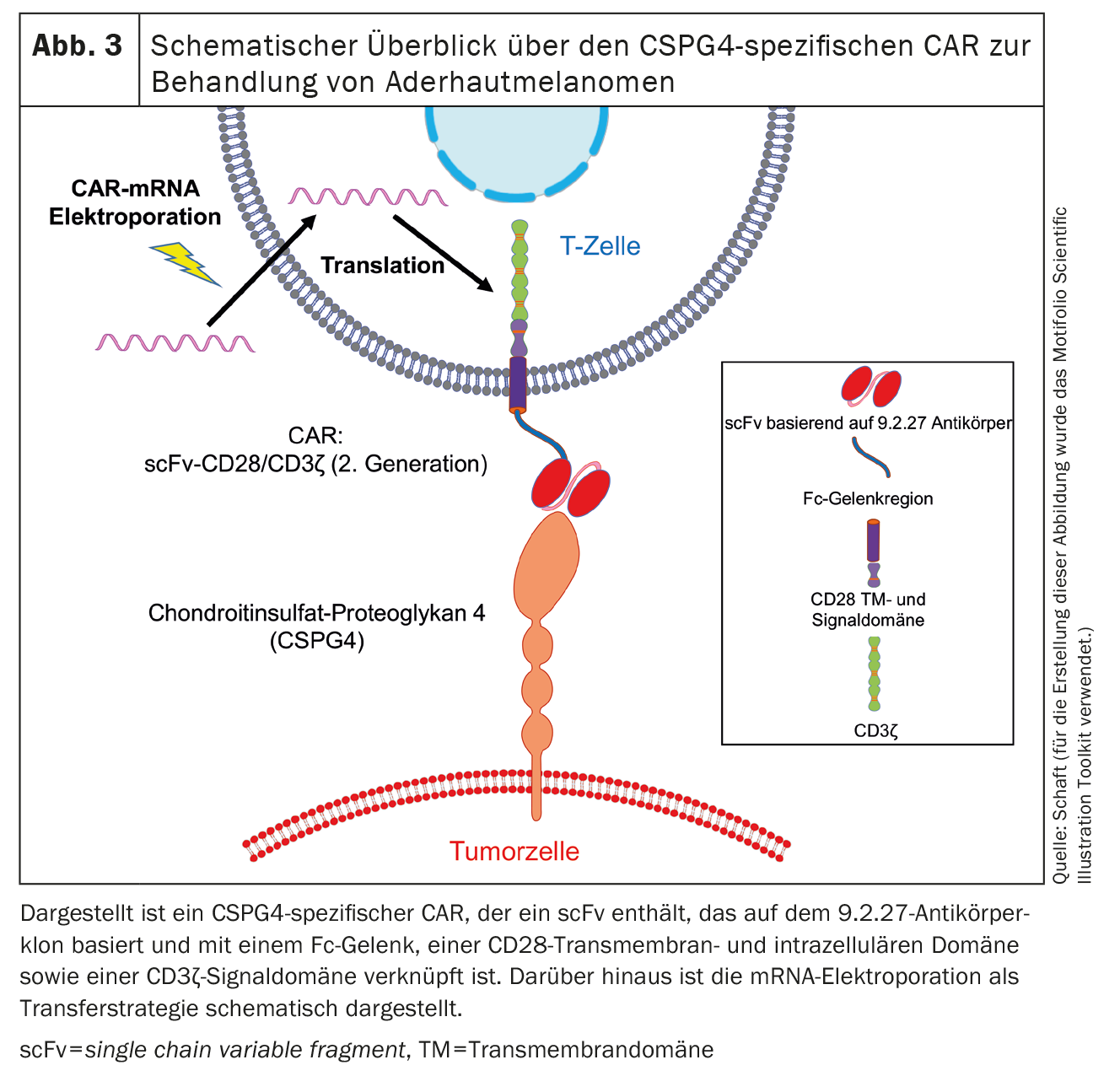
Il secondo antigene espresso nei melanomi uveali è il condroitin-solfato proteoglicano 4 (CSPG4) (Fig. 3), precedentemente noto anche come condroitin-solfato proteoglicano associato al melanoma (MCSP) o antigene associato al melanoma ad alto peso molecolare (HMW-MAA). La CSPG4 è una proteina transmembrana a passaggio singolo di tipo 1 ed è stata scoperta da Ralph Reisfeld [72]. Noi e altri gruppi di lavoro [47-50, 62-71] abbiamo lavorato principalmente sul CSPG4. L’espressione di CSPG4 è associata a una maggiore proliferazione e a una maggiore sopravvivenza delle cellule tumorali. Ciò viene avviato dall’attivazione della via di segnalazione MAPK e dalla rappresentazione incrociata dei fattori di crescita [73]. Inoltre, CSPG4 svolge un ruolo nella motilità cellulare e nell’infiltrazione tissutale grazie alla sua associazione con il citoscheletro di actina e al legame con varie integrine e componenti della matrice extracellulare [74]. Inoltre, CSPG4 è coinvolto nella formazione della placenta [75], nell’angiogenesi [76], nella formazione di reti neuronali [77], nel turnover dei cheratinociti e nell’omeostasi delle cellule staminali epidermiche [78].
Diverse pubblicazioni hanno descritto l’espressione di CSPG4 su tessuti non patologici, come i precursori del follicolo pilifero e le cellule epidermiche, le cellule endoteliali e i periciti attivati (ma non sui vasi maturi) [79,80], i condrociti della cartilagine articolare [81], le cellule muscolari lisce [82] e le cellule della sinapsi neuromuscolare del muscolo scheletrico umano postnatale [83]. Tuttavia, la CSPG4 è espressa in misura significativamente minore nei tessuti sani rispetto alle cellule tumorali [62,73,84].
Beard et al. hanno dimostrato che CSPG4 può essere rilevato a livello di RNA in una varietà di tessuti normali, tra cui il sistema nervoso centrale, l’occhio, la pelle, il tessuto adiposo, i vasi sanguigni, la vescica, il tratto gastrointestinale, l’utero, la prostata, la milza e il timo [85]. In media, l’RNA di CSPG4 è sovraespresso 6,6 volte nel tessuto maligno (melanoma) rispetto al tessuto sano [85]. Questi risultati hanno confermato il lavoro precedente di Erfurt et al. che ha dimostrato che, sebbene l’mRNA di CSPG4 potesse essere rilevato in alcuni campioni di tessuto normale, l’espressione era significativamente aumentata nei campioni di melanoma cutaneo e di melanoma uveale [86].
La colorazione immunoistochimica e gli array di proteine in fase inversa hanno mostrato che l’espressione specifica di CSPG4 a livello proteico poteva essere rilevata solo in alcuni campioni dell’intestino tenue [63]. Non è stata rilevata l’espressione della proteina CSPG4 nei seguenti tessuti: Cervello, nervi periferici, pelle, mesotelio, seno, cuore, rene, ghiandole surrenali, fegato, polmone, linfonodi, muscoli, ovaie, pancreas, esofago, prostata, milza, stomaco, utero e tiroide [62,63]. Al contrario, CSPG4 è espresso in quasi tutte le cellule di melanoma cutaneo [87–90]. I melanomi coroideali [91,92] e alcuni altri tumori come i sarcomi, gli astrocitomi, i gliomi, i neuroblastomi [93-96], le leucemie [97–101] e il cancro al seno triplo negativo esprimono anche la CSPG4 [102]. In molti di questi tumori maligni, l’espressione di CSPG4 è associata a una prognosi sfavorevole e a una crescita tumorale aggressiva [103].
Inoltre, CSPG4 è considerato un antigene bersaglio primario del tumore [84], poiché svolge un ruolo nella metastasi del melanoma [104] ed è espresso sui periciti attivati durante l’angiogenesi nei tumori e nell’ipossia [105–107]. Quest’ultima permette di mirare alla vascolarizzazione del tumore. Soprattutto, CSPG4 agisce come driver oncogeno nel melanoma e promuove la crescita e la sopravvivenza delle cellule maligne dopo l’attivazione di varie vie di segnalazione [73]. Pertanto, il tumore non può semplicemente downregolare CSPG4 per sfuggire alla terapia mirata a CSPG4.
Per questo motivo, CSPG4 è già stato selezionato come antigene bersaglio da diversi gruppi e i CAR specifici per CSPG4 sono stati introdotti nelle cellule T attraverso vari meccanismi. I CAR specifici per CSPG4 di diversi formati, trasdotti viralmente nelle cellule T, hanno determinato una forte citotossicità delle cellule T in vitro . Nei modelli animali, le cellule T trasferite adottivamente hanno reagito a vari tumori che esprimono CSPG4, come il melanoma, il cancro al seno, il mesotelioma, il glioblastoma e l’osteosarcoma [47–50,62–71]. Geldres et al. hanno trasdotto retroviralmente un CAR di seconda generazione specifico per CSPG4 in cellule T. In vitro, queste cellule T CAR specifiche per CSPG4 sono state in grado di riconoscere e lisciare le cellule di melanoma in modo antigene-specifico [65]. Inoltre, il legame dell’antigene con il CAR ha portato a una marcata secrezione di IL-2 e IFNγ. In vivo , il trasferimento di cellule CAR-T specifiche per CSPG4 in topi portatori di cellule di melanoma ha portato a un rallentamento significativo della crescita tumorale e a un miglioramento della sopravvivenza complessiva dei topi [65]. Nella stessa pubblicazione, è stata descritta la mancanza di reattività positiva delle cellule CAR-T specifiche per CSPG4 verso il tessuto normale con RNA CSPG4 rilevabile ma senza espressione della proteina CSPG4 [65]. Hanno potuto dimostrare che le cellule CAR-T specifiche per CSPG4 non hanno esercitato una citotossicità significativa contro le linee cellulari epiteliali primarie della prostata, del polmone e del rene [65]. Pertanto, le analisi di espressione di Beard et al. [63,85] mitigano le preoccupazioni descritte sopra in merito allatossicità on-target/off-tumour indotta dalle cellule CAR-T specifiche di CSPG4, in quanto l’espressione di CSPG4 a livello proteico è necessaria per indurre una reattività indesiderata delle cellule CAR-T.
Finora, abbiamo utilizzato il metodo di trasfezione dell’mRNA mediante elettroporazione per introdurre i CAR nelle cellule T. Nel corso di questo lavoro, abbiamo già testato diversi CAR specifici per CSPG4 e abbiamo osservato che le cellule CAR-T trasfettate con mRNA sono in grado di eliminare le cellule tumorali in modo antigene-specifico. La cinetica di espressione delle CAR trasferite tramite elettroporazione dipende dal backbone della CAR [47]. È stato identificato un CAR che presenta un’elevata espressione sulle cellule T e un’alta reattività anti-tumorale. Questo CAR contiene un scFv basato sul clone dell’anticorpo 9.2.27 collegato a uno spaziatore Fc, un dominio transmembrana e intracellulare CD28 e un dominio di segnalazione CD3ζ. [47](Fig. 3). Esperimenti in vivo con topi immunodeficienti Rag-/-/ con catena γ comune-/- hanno dimostrato che le cellule CAR-T trasfettate specifiche per CSPG4 hanno prolungato significativamente il tempo medio di sopravvivenza dei topi [47].
Per trasferire le cellule CAR-T specifiche per CSPG4 nell’applicazione clinica, la produzione di cellule CAR-T su scala clinica è stata stabilita in laboratorio mediante la trasfezione di mRNA di un CAR in piena conformità alle GMP [50]. Questo ha dimostrato che è possibile la produzione ripetuta di un numero sufficiente di cellule T CSPG4-specifiche CAR-trasfettate altamente pure. Queste cellule T modificate mostrano un’efficienza di trasfezione molto elevata, un’alta espressione di CAR e un’elevata efficacia nell’uccidere le cellule bersaglio del melanoma [50].
Sebbene CSPG4 sia un antigene target tumorale primario, in particolare nel melanoma cutaneo, abbiamo anche scoperto che non era espresso in diverse linee cellulari di melanoma uveale che abbiamo analizzato. Abbiamo quindi creato una piattaforma combinata in silico/in vitro per identificare nuovi antigeni della superficie cellulare specifici del tumore dei melanomi uveali. Utilizzando questa piattaforma, abbiamo identificato una proteina candidata come antigene target adatto nel melanoma uveale per lo sviluppo di ulteriori CAR. I CAR generati in grado di legarsi a questa proteina candidata sono attualmente in fase di test per verificarne la funzionalità e la specificità.
Prospettiva
L’uso delle cellule CAR-T nel trattamento dei tumori solidi ha un grande potenziale. Sono necessari ulteriori studi preclinici e sperimentazioni cliniche per rispondere all’elevata esigenza medica di trattamento delle entità tumorali solide (come il melanoma uveale).
I futuri studi clinici dovrebbero concentrarsi sulla sperimentazione di nuovi formati CAR. Oltre a testare nuovi domini extracellulari di legame con l’antigene e nuovi domini di segnalazione intracellulare [109], questo include anche la sperimentazione di formati che aumentano la sicurezza dell’uso delle cellule CAR-T [108]. Inoltre, i nuovi veicoli cellulari per il trasferimento CAR [109,110] promettono di ampliare la gamma di applicazioni. Per esempio, la possibilità di utilizzare le cellule CAR-NK [111] o le cellule CAR-T allogeniche [112] come standard può ridurre i costi della terapia cellulare CAR e quindi rendere la terapia accessibile a più pazienti.
Inoltre, è necessario trovare più antigeni specifici per il tumore, per evitare reazioni on-target/off-tumour. Gli antigeni che sono espressi nello stroma tumorale e che possono essere attaccati dalle cellule CAR-T sono molto promettenti in questo settore [113]. Il targeting di più antigeni da parte di una cellula CAR-T (cioè l’espressione di diversi CAR specifici per antigeni diversi su una singola cellula) può aumentare la specificità tumorale e ridurre il rischio di effetti fuori bersaglio. Questo vale anche per il modello in cui i moduli di segnalazione intracellulare sono divisi tra i diversi CAR, al fine di aumentare il profilo di sicurezza delle cellule CAR-T. Questo rende anche meno probabile lo sviluppo di varianti di perdita di antigene dei tumori.
Inoltre, le terapie di combinazione delle cellule CAR-T con varie piccole molecole o anticorpi monoclonali per prevenire i meccanismi di fuga del tumore e aumentare l’attività antitumorale sono già in fase di sperimentazione clinica in molti tumori ematologici (panoramica dettagliata in [114,115]). Tali combinazioni sono promettenti anche per il trattamento dei tumori solidi e devono essere testate in studi clinici nel prossimo futuro.
N.S. ha condotto la ricerca su clinicaltrials.gov, ha rivisto le figure e ha redatto il manoscritto. S.H. ha redatto il manoscritto. N.S. e S.H. hanno redatto insieme le domande di formazione ECM.
Letteratura:
- Gross G, Gorochov G, Waks T, Eshhar Z: Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc 1989; 21(1 Pt 1): 127–130.
- Gross G, Waks T, Eshhar Z: Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989; 86(24): 10024–10028.
- June CH, Sadelain M: Chimeric Antigen Receptor Therapy. N Engl J Med 2018; 379(1): 64–73.
- Maude SL, Laetsch TW, Buechner J, et al.: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018; 378(5): 439-448.
- Neelapu SS, Locke FL, Go WY: CAR T-Cell Therapy in Large B-Cell Lymphoma. N Engl J Med 2018; 378(11): 1065.
- Schuster SJ, Bishop MR, Tam CS, et al.: Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019; 380(1): 45–56.
- Wang Z, Guo Y, Han W: Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017; 8(12): 896–925.
- Han S, Latchoumanin O, Wu G, et al.: Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett 2017; 390: 188–200.
- Yeku O, Li X, Brentjens RJ: Adoptive T-Cell Therapy for Solid Tumors. Am Soc Clin Oncol Educ Book 2017; 37: 193–204.
- Arabi F, Torabi-Rahvar M, Shariati A, et al.: Antigenic targets of CAR T Cell Therapy. A retrospective view on clinical trials. Exp Cell Res 2018; 369(1): 1–10.
- Schaft N: The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers (Basel) 2020; 12(9).
- Holzinger A, Abken H: CAR T Cells: A Snapshot on the Growing Options to Design a CAR. Hemasphere 2019; 3(1): e172.
- Boomer JS, Green JM: An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol 2010; 2(8): a002436.
- Kawalekar OU, O’Connor RS, Fraietta JA, et al.: Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016; 44(2): 380–390.
- Cannons JL, Choi Y, Watts TH: Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol 2000; 165(11): 6193–6204.
- Lee HW, Nam KO, Park SJ, Kwon BS: 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol 2003; 33(8): 2133–2141.
- Zhao Z, Condomines M, van der Stegen SJC, et al.: Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015; 28(4): 415–428.
- Milone MC, Fish JD, Carpenito C, et al.: Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17(8): 1453–1464.
- Lim WA, June CH: The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017; 168(4): 724–740.
- Roselli E, Frieling JS, Thorner K, et al.: CAR-T Engineering: Optimizing Signal Transduction and Effector Mechanisms. BioDrugs 2019; 33(6): 647–659.
- Hombach AA, Holzinger A, Abken H.: The weal and woe of costimulation in the adoptive therapy of cancer with chimeric antigen receptor (CAR)-redirected T cells. Curr Mol Med 2013; 13(7): 1079–1088.
- Sadelain M, Brentjens R, Riviere I: The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3(4): 388-398.
- Redeker A, Arens R: Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front Immunol 2016; 7: 345.
- Weinkove R, George P, Dasyam N, McLellan AD.: Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology 2019; 8(5): e1049.
- Ceppi F, Rivers J, Annesley C, et al.: Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion 2018; 58(6): 1414–1420.
- Li W, Guo L, Rathi P, et al.: Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum Gene Ther 2017; 28(5): 437–448.
- Chmielewski M, Hombach AA, Abken H: Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257(1): 83–90.
- Chmielewski M, Kopecky C, Hombach AA, Abken H: IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71(17): 5697–5706.
- Pegram HJ, Lee JC, Hayman EG, et al.: Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012; 119(18): 4133-4141.
- Koneru M, Purdon TJ, Spriggs D, et al.: IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015; 4(3): e994446.
- Koneru M, O›Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ.: A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 2015; 13: 102.
- Xu A, Bhanumathy KK, Wu J, et al.: IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci 2016; 6: 30.
- Lamers CH, Willemsen R, van Elzakker P, et al.: Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011; 117(1): 72–82.
- Magnani CF, Tettamanti S, Alberti G, et al.: Transposon-Based CAR T Cells in Acute Leukemias: Where are We Going? Cells 2020; 9(6).
- Hudecek M, Ivics Z: Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr Opin Genet Dev 2018; 52: 100–108.
- Tipanee J, VandenDriessche T, Chuah MK: Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum Gene Ther 2017; 28(11): 1087–1104.
- Vargas JE, Chicaybam L, Stein RT, et al.: Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med 2016; 14(1): 288.
- Ran FA, Hsu PD, Wright J, et al.: Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8(11): 2281–2308.
- Birkholz K, Hombach A, Krug C, et al.: Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther 2009; 16(5): 596–604.
- Lamers CH, Sleijfer S, et al.: Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21(4): 904–912.
- Morgan RA, Yang JC, Kitano M, et al.: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843-851.
- Stavrou M, Philip B, Traynor-White C, et al.: A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol Ther 2018; 26(5): 1266–1276.
- Di Stasi A, Tey SK, Dotti G, et al.: Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365(18): 1673–1683.
- Moolten FL: Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986; 46(10): 5276–5281.
- Beltinger C, Fulda S, Kammertoens T, et al.: Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci U S A 1999; 96(15): 8699–8704.
- Harrer DC, Simon B, Fujii SI, et al.: RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17(1): 551.
- Krug C, Birkholz K, Paulus A, et al.: Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol Immunother 2015; 64(12): 1623–1635.
- Dörrie J, Babalija L, Hoyer S, et al.: BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int J Mol Sci 2018; 19(1).
- Harrer DC, Schuler G, Dörrie J, Schaft N: CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int J Mol Sci 2019; 20(11).
- Wiesinger M, Marz J, Kummer M, et al.: Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers (Basel) 2019; 11(8).
- Shi J, Ma Y, Zhu J, et al.: A Review on Electroporation-Based Intracellular Delivery. Molecules 2018; 23(11).
- Ernst M, Oeser A, Besiroglu B, et al.: Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst Rev 2021; 9(9): Cd013365.
- Pinc A, Somasundaram R, Wagner C, et al.: Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol Ther 2012; 20(5): 1056–1062.
- Schmidt P, Kopecky C, Hombach A, et al.: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474–2479.
- Brignole C, Marimpietri D, Pagnan G, et al.: Neuroblastoma targeting by c-myb-selective antisense oligonucleotides entrapped in anti-GD2 immunoliposome: immune cell-mediated anti-tumor activities. Cancer Lett 2005; 228(1–2): 181–186.
- Mitri Z, Constantine T, O’Regan R: The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res Pract 2012; 2012: 743193.
- Slamon DJ, Godolphin W, Jones LA, et al.: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244(4905): 707–712.
- Zhang JG, Kruse CA, Driggers L, et al.: Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol 2008; 88(1): 65–76.
- Casucci M, Hawkins RE, Dotti G, Bondanza A: Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother 2015; 64(1): 123–130.
- Morgan RA, Yang JC, Kitano M, et al.: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843–851.
- Slamon DJ, Leyland-Jones B, Shak S, et al.: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344(11): 783–792.
- Wang Y, Geldres C, Ferrone S, Dotti G: Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin Ther Targets 2015; 19(10): 1339–1350.
- Beard RE, Zheng Z, Lagisetty KH, et al.: Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer 2014; 2: 25.
- Pellegatta S, Savoldo B, Di IN, et al.: Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med 2018; 10(430).
- Geldres C, Savoldo B, Hoyos V, et al.: T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res 2014; 20(4): 962–971.
- Abken H, Hombach A, Heuser C, Reinhold U: A novel strategy in the elimination of disseminated melanoma cells: chimeric receptors endow T cells with tumor specificity. Recent Results Cancer Res 2001; 158: 249–264.
- Burns WR, Zhao Y, Frankel TL, et al.: A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res 2010; 70(8): 3027–3033.
- Losch FO, Muller R, Mutschler B, et al.: Activation of T cells via tumor antigen specific chimeric receptors: the role of the intracellular signaling domain. Int J Cancer 2003; 103(3): 399–407.
- Reinhold U, Liu L, Ludtke-Handjery HC, et al.: Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J Invest Dermatol 1999; 112(5): 744–750.
- Schmidt P, Kopecky C, Hombach A, et al.: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474-2479.
- Harrer D, Simon B, Fujii SI, et al.: RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17: 551.
- Bumol TF, Reisfeld RA.: Unique glycoprotein-proteoglycan complex defined by monoclonal antibody on human melanoma cells. Proc Natl Acad Sci USA 1982; 79(4): 1245–1249.
- Ilieva KM, Cheung A, Mele S, et al.: Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front Immunol 2017; 8: 1911.
- Schiffer D, Mellai M, Boldorini R, et al.: The Significance of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Human Gliomas. Int J Mol Sci 2018; 19(9).
- Van Sinderen M, Cuman C, Winship A, et al.: The chrondroitin sulfate proteoglycan (CSPG4) regulates human trophoblast function. Placenta 2013; 34(10): 907–912.
- Fukushi J, Makagiansar IT, Stallcup WB: NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell 2004; 15(8): 3580–3590.
- Sakry D, Neitz A, Singh J, et al.: Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol 2014; 12(11): e1001993.
- Legg J, Jensen UB, Broad S, et al.: Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003; 130(24): 6049–6063.
- Ferrone S, Chen ZJ, Liu CC, et al.: Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharmacol Ther 1993; 57(2-3): 259–290.
- Schlingemann RO, Rietveld FJ, de Waal RM, et al.: Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol 1990; 136(6): 1393–1405.
- Midwood KS, Salter DM: Expression of NG2/human melanoma proteoglycan in human adult articular chondrocytes. Osteoarthritis Cartilage 1998; 6(5): 297–305.
- Tordsson JM, Ohlsson LG, Abrahmsen LB, et al.: Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol Immunother 2000; 48(12): 691–702.
- Petrini S, Tessa A, Carrozzo R, et al.: Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol Cell Neurosci 2003; 23(2): 219–231.
- Campoli MR, Chang CC, Kageshita T, et al.: Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol 2004; 24(4): 267–296.
- Beard RE, Abate-Daga D, Rosati SF, et al.: Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin Cancer Res 2013; 19(18): 4941–4950.
- Erfurt C, Sun Z, Haendle I, et al.: Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J Immunol 2007; 178(12): 7703–7709.
- Natali PG, Giacomini P, Russo C, et al.: Antigenic profile of human melanoma cells. Analysis with monoclonal antibodies to histocompatibility antigens and to melanoma-associated antigens. J Cutan Pathol 1983; 10(4): 225–237.
- Berd D, Herlyn M, Koprowski H, Mastrangelo MJ: Flow cytometric determination of the frequency and heterogeneity of expression of human melanoma-associated antigens. Cancer Res 1989; 49(23): 6840–6844.
- Morgan AC Jr., Galloway DR, Reisfeld RA: Production and characterization of monoclonal antibody to a melanoma specific glycoprotein. Hybridoma 1981; 1(1): 27–36.
- Morgan AC Jr., Woodhouse C, Bartholemew R, Schroff R: Human melanoma-associated antigens: analysis of antigenic heterogeneity by molecular, serologic and flow-cytometric approaches. Mol Immunol 1986; 23(2): 193–200.
- Li Y, Madigan MC, Lai K, et al.: Human uveal melanoma expresses NG2 immunoreactivity. Br J Ophthalmol 2003; 87(5): 629–632.
- Li Y, Wang J, Rizvi SM, Jager MJ, et al.: In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest Ophthalmol Vis Sci 2005; 46(12): 4365–4371.
- Chekenya M, Rooprai HK, Davies D, et al.: The NG2 chondroitin sulfate proteoglycan: role in malignant progression of human brain tumours. Int J Dev Neurosci 1999; 17(5–6): 421–435.
- Godal A, Bruland O, Haug E, et al.: Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br J Cancer 1986; 53(6): 839–841.
- Shoshan Y, Nishiyama A, Chang A, et al.: Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci U S A 1999; 96(18): 10361–10366.
- Yadavilli S, Hwang EI, Packer RJ, Nazarian J: The Role of NG2 Proteoglycan in Glioma. Transl Oncol 2016; 9(1): 57–63.
- Behm FG, Smith FO, Raimondi SC, et al.: Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996; 87(3): 1134–1139.
- Hilden JM, Smith FO, Frestedt JL, et al.: MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997; 89(10): 3801–3805.
- Schwartz S, Rieder H, Schlager B, et al.: Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia 2003; 17(8): 1589–1595.
- Smith FO, Rauch C, Williams DE, et al.: The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996; 87(3): 1123–1133.
- Wuchter C, Harbott J, Schoch C, et al.: Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000; 14(7): 1232–1238.
- Wang X, Osada T, Wang Y, et al.: CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst 2010; 102(19): 1496–1512.
- Nicolosi PA, Dallatomasina A, Perris R: Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015; 5(5): 530–544.
- de Vries JE, Keizer GD, te Velde AA, et al.: Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int J Cancer 1986; 38(4): 465–473.
- Ozerdem U: Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res 2006; 38(5): 251–254.
- Ozerdem U: Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 2006; 66(3): 294–304.
- Ampofo E, Schmitt BM, Menger MD, Laschke MW: The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol Biol Lett 2017; 22: 4.
- Stoiber S, Cadilha BL, Benmebarek MR, et al.: Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019; 8(5).
- Sievers NM, Dörrie J, Schaft N: CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int J Mol Sci 2020; 21(10).
- Harrer DC, Dörrie J, Schaft N: Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum Gene Ther 2018; 29(5): 547–558.
- Montagner IM, Penna A, Fracasso G, et al.: Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells 2020; 9(6).
- Van Cutsem E, Machiels J, Van den Eynde M, et al.: SO-009 – Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Annals of Oncology 2019; 30: iv124–iv125.
- Santoro SP, Kim S, Motz GT, et al.: T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res 2015; 3(1): 68–84.
- Bansal R, Reshef R: Revving the CAR – Combination strategies to enhance CAR T cell effectiveness. Blood Reviews 2020: 100695.
- Harrer DC, Dorrie J, Schaft N: CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int J Mol Sci 2023; 24(3).
DERMATOLOGIE PRAXIS 2024; 19(1): 14–24









