
Se si sospetta un tumore secernente catecolamine, la determinazione delle metanefrine nel plasma libero è un metodo diagnostico sensibile. L’ipertensione parossistica è un sintomo comune. A seconda dei risultati di laboratorio, è necessario eseguire un esame di imaging per localizzare il tumore. Per i tumori non resecabili o metastatici, esistono nuove terapie sperimentali basate sulla firma genetica e molecolare individuale, in cui si ripongono molte speranze. In questo contesto, le analisi genetiche stanno acquisendo sempre più importanza.
I feocromocitomi (PCC) sono tumori che producono catecolamine e che hanno origine dalle cellule cromaffini della midollare surrenale. I paragangliomi (PGL) sono tumori che producono noradrenalina e dopamina nei paragangliomi simpatici e parasimpatici [1]. Il quadro clinico e la procedura diagnostica iniziale sono molto simili in entrambi i tipi di tumore secernenti catecolamine [2]. Un sintomo tipico del feocromocitoma è l’ipertensione, che si manifesta in modo parossistico in circa la metà dei pazienti affetti. Altri segni della malattia includono sudorazione, palpitazioni, mal di testa, tremore o nausea. Nella maggior parte dei casi, i PGL sono facilmente curabili con una resezione completa, ma ci sono differenze significative nel rischio di malignità a seconda del genotipo e della posizione del tumore. I tumori che metastatizzano sono considerati maligni, ossia circa il 10-15% di tutti i PCC e il 35-40% di tutti i PGL [1]. Tutti i pazienti con feocromocitomi/paragangliomi (PPGL) dovrebbero sottoporsi a test genetici, ha sottolineato la Prof.ssa Dr. med. Svenja Nölting, medico senior della Clinica di Endocrinologia, Diabetologia e Nutrizione Clinica, Ospedale Universitario di Zurigo, in occasione del meeting annuale della Società Svizzera di Endocrinologia e Diabetologia (SGED) lo scorso novembre [1].

Tipizzazione dei cluster: firma genetica e molecolare
“Circa il 70% di tutte le PPGL può essere assegnato a uno dei tre cluster molecolari in base alla loro mutazione di base”, ha spiegato il relatore [1,3]. La maggior parte dei tumori appartiene ai cluster 1 e 2; del cluster 3 si sa ancora poco. I tumori del cluster 1 si basano sull’attivazione delle vie di segnalazione della pseudoipossia, quelli del cluster 2 sull’attivazione delle vie di segnalazione dipendenti dalla tirosina chinasi [4]. Nel 30-35% dei casi, sono presenti mutazioni germinali ereditate in modo autosomico-dominante, mentre il 35-40% è dovuto a mutazioni somatiche. I tumori del cluster 1 sono più spesso localizzati a livello extra-adrenale e presentano il rischio più elevato di metastasi, mentre i tumori del cluster 2 sono per lo più localizzati a livello surrenale e presentano un basso rischio di metastasi [4].
Diagnostica: le metanefrine nel plasma libero sono marcatori significativi
I pazienti con mutazione germinale nota, ma anche con sospetto clinico di feocromocitoma/paraganglioma (PPGL) o se è stata riportata una storia di incidentaloma surrenalico o di PPGL, devono essere sottoposti a screening [4]. In una prima fase, devono essere raccolti i seguenti parametri biochimici di laboratorio [1]:
- Metanefrine nel plasma libero (=metaboliti dell’adrenalina)
- Normetanefrine (=metaboliti della noradrenalina)
- 3-metossi-tiramina (=metaboliti della dopamina)
È consigliabile raccogliere questi dati utilizzando la spettrometria di massa, in quanto può raggiungere una sensibilità migliore rispetto ad altri metodi. Prima del prelievo del sangue, i pazienti devono astenersi dalla nicotina e dal caffè, nonché dall’assumere SSRI e/o antidepressivi triciclici, metanfetamine o cocaina, tra le altre cose. Oltre ai parametri citati, è utile anche determinare la cromogranina A, in quanto si tratta di un buon marcatore, soprattutto per i tumori asintomatici, per cui è necessario astenersi dall’assunzione di inibitori della pompa protonica (PPI) una settimana prima dell’esame. La determinazione del catecolo da sola non è utile, ha detto, perché potrebbe portare a un risultato falso-negativo. Se i valori di metanefrina determinati sono superiori al doppio del valore normale, la diagnostica per immagini è indicata in un’ulteriore fase di chiarimento [1]. Si raccomanda innanzitutto la risonanza magnetica/TC dell’addome e della regione pelvica; se i risultati sono negativi o se c’è già un tumore extra-adrenale/PGL, è necessaria l’imaging funzionale. Se le dimensioni del tumore surrenale sono <5 cm, Prof. Nölting consiglia l’uso della DOPA PET/CT. La DOTA-SSA-PET/CT deve essere eseguita in presenza di tumori extra-adrenali e di risultati di imaging anatomico negativi o se sono già presenti metastasi o le dimensioni del tumore surrenale sono >5 cm, ha spiegato il relatore [1].
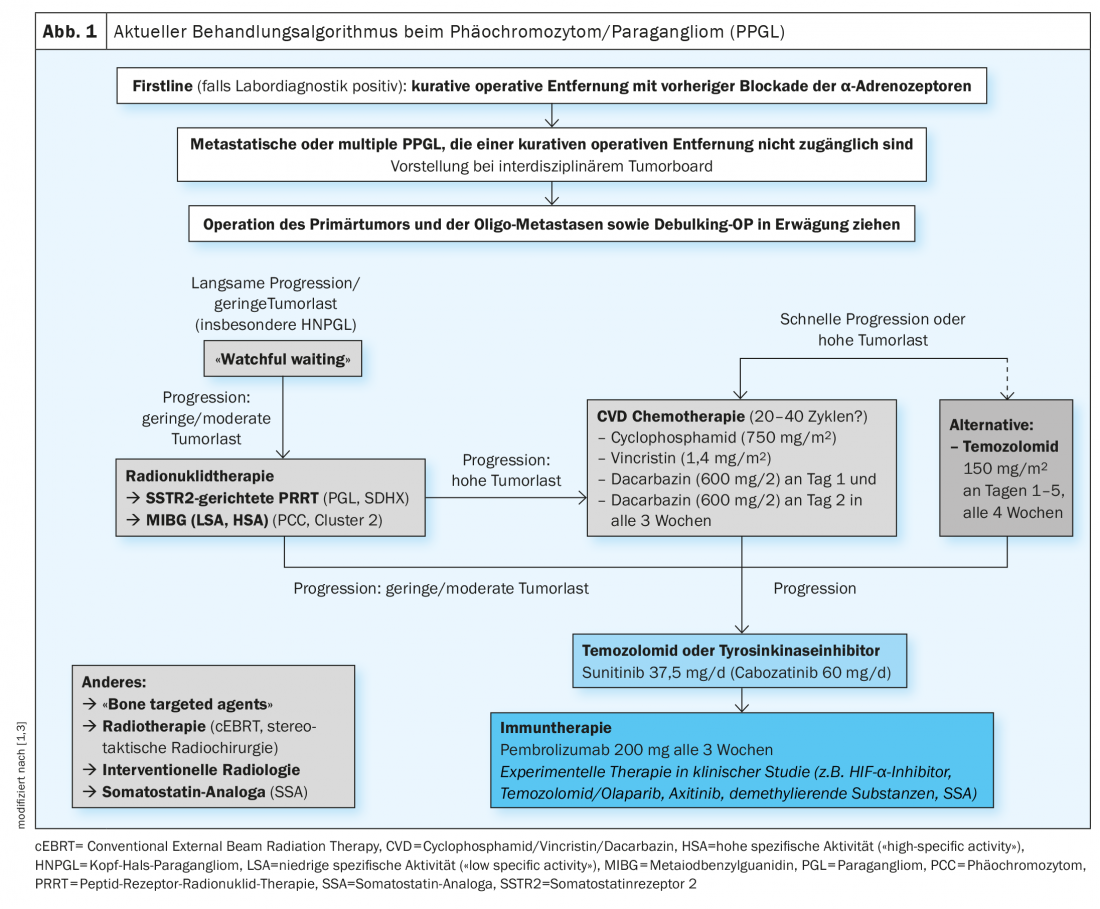
Strategia di trattamento individualizzata: nuove terapie sperimentali
L’attuale algoritmo di trattamento è mostrato nella Figura 1 [1,3]. La rimozione chirurgica è considerata la terapia di prima linea per il PPGL localizzato. Di solito può essere eseguita in modo minimamente invasivo e deve essere eseguita in un centro esperto [4]. Per evitare complicazioni, viene effettuato preoperatoriamente un blocco medicinale degli α-adrenocettori. Il PPGL metastatico viene attualmente trattato con la terapia con radionuclidi, la chemioterapia o gli inibitori della tirosin-chinasi, anche se non esistono ancora terapie ufficialmente approvate. Altre nuove strategie di trattamento sono attualmente in fase di sperimentazione clinica. Finora, la terapia cluster-specifica della malattia inoperabile o metastatica non è stata ancora stabilita nella pratica clinica. Tuttavia, questo avrebbe senso per poter offrire un trattamento personalizzato e geneticamente controllato, afferma il Prof. Nölting [3]. Una pubblicazione scientifica è apparsa su Endocrine Reviews nel 2021, in cui si discute in dettaglio di come si possa sviluppare un piano di gestione del paziente coerente e individualizzato per i pazienti con feocromocitoma/paraganglioma, basato sulle analisi genetiche e sulla tipizzazione dei cluster, rispettivamente, al fine di adattare in modo ottimale le opzioni di trattamento [3].
Congresso: Società Svizzera di Endocrinologia e Diabetologia 11.11.2021
Letteratura:
- Nölting S: “Test genetici nei pazienti con feocromocitomi/paragangliomi”, Prof. Dr med. Svenja Nölting, SGED 11.11.2021
- Zulewski H, Grouzmann E: Diagnosi e trattamento: “Feocromocitoma”, Swiss Med Forum 2017; 17(37): 790-796, https://medicalforum.ch/de/detail/doi/smf.2017.03057
- Nölting S, et al: Gestione personalizzata del feocromocitoma e del paraganglioma. Endocr Rev 2021 Jun 19:bnab019
- Feocromocitoma – malattia modello per la medicina personalizzata, Dtsch Med Wochenschr 2021; 146(23): 1520-1526.
- Società tedesca di endocrinologia, ormoni e metabolismo: PROSPHEO, www.endokrinologie.net/sektion-nebenniere-steroide-hypertonie-7.php (ultimo accesso 07.12.2021).
PRATICA GP 2022; 17(1): 42-43
InFo ONCOLOGIA & EMATOLOGIA 2022; 10(3): 18-19









