I miglioramenti nella gestione della malattia aumentano la qualità e l’aspettativa di vita dei pazienti con fibrosi cistica. Lo screening neonatale introdotto nel 2011 consente una diagnosi precoce. Tuttavia, la malattia deve essere considerata anche come una possibile diagnosi differenziale in età adulta.
La fibrosi cistica (CF, a volte anche fibrosi cistica) è la malattia metabolica congenita più comune, che in ultima analisi limita la vita, in Svizzera. Come malattia sistemica, la FC colpisce principalmente la funzione delle ghiandole esocrine del tratto respiratorio e del tratto digestivo. In particolare, il decorso polmonare con distruzione continua e irreversibile dell’organo causa un’aspettativa di vita ridotta. Grazie al miglioramento delle misure mediche, i soggetti colpiti raggiungono oggi l’età adulta quasi senza eccezioni, ma rappresentano ancora un collettivo relativamente nuovo nella pratica del medico di famiglia. Come innovazione promettente, dall’inizio del 2014 è disponibile una terapia causale per una percentuale molto ridotta di pazienti.
Modello di malattia
Già nel 1936, la fibrosi cistica, che all’epoca aveva ancora un nome diverso, era stata descritta dal pediatra zurighese Guido Fanconi come una malattia mortale dei bambini piccoli, motivo per cui l’accesso a questa patologia rimase precluso ai colleghi della medicina di famiglia per molto tempo.
La fibrosi cistica è la malattia ereditaria congenita più comune, cronica e, in ultima analisi, limitante la vita, con una prevalenza di circa 1:2500. Circa una persona su 25 in Europa centrale è portatrice sana di una mutazione. Ci sono circa 70.000 persone affette da questa malattia in tutto il mondo, e si stima che in Svizzera ce ne siano circa 900. Circa la metà dei pazienti ha più di 18 anni. La modalità di ereditarietà è autosomica recessiva, il che significa che statisticamente un bambino su quattro nato da una relazione tra due portatori sani di mutazione è affetto da FC, due bambini sono anch’essi portatori sani di mutazione come i loro genitori, e un bambino non è né portatore di mutazione né malato. Nel 1989, il gene sottostante è stato identificato sul cromosoma 7. La causa è una mutazione nel gene CFTR (“regolatore di conduttanza transmembrana della fibrosi cistica”), che codifica per il canale del cloruro nella membrana cellulare [1,2].
Clinica
Nella malattia sistemica, sono colpiti diversi organi, ma di solito la malattia polmonare cronica comporta un aumento della morbilità e una riduzione dell’aspettativa di vita. Grazie al miglioramento delle possibilità mediche, l’aspettativa di vita è aumentata costantemente negli ultimi anni. L’aspettativa di vita media in Europa è attualmente superiore a 40 anni e continuerà ad aumentare, soprattutto nella generazione rilevata dallo screening neonatale in Svizzera dal 2011. Tuttavia, il decorso della malattia è molto incostante e alcuni pazienti muoiono ancora di insufficienza polmonare in giovane età. Comune a tutti i malati sono le infezioni batteriche croniche del tratto respiratorio, che portano alla distruzione irreversibile dei polmoni. I pazienti soffrono di tosse, espettorato e di una crescente limitazione della loro resistenza fisica. Inoltre, la diarrea cronica con maldigestione è dovuta all’insufficienza pancreatica esocrina, che colpisce circa l’85% dei pazienti. Come conseguenza della malnutrizione, ma anche dell’aumento del lavoro respiratorio, si verifica una mancata crescita. Circa il 10% presenta già ileo da meconio dopo la nascita, ma anche le persone più anziane possono soffrire di sindromi da ostruzione intestinale ricorrente. Nonostante la terapia sintomatica ottimale, nel prosieguo della malattia possono verificarsi anche cirrosi epatica o insufficienza endocrina del pancreas con sviluppo di diabete mellito. I pazienti di sesso maschile con FC, in particolare, sono spesso affetti da infertilità [1,2].
Fisiopatologia
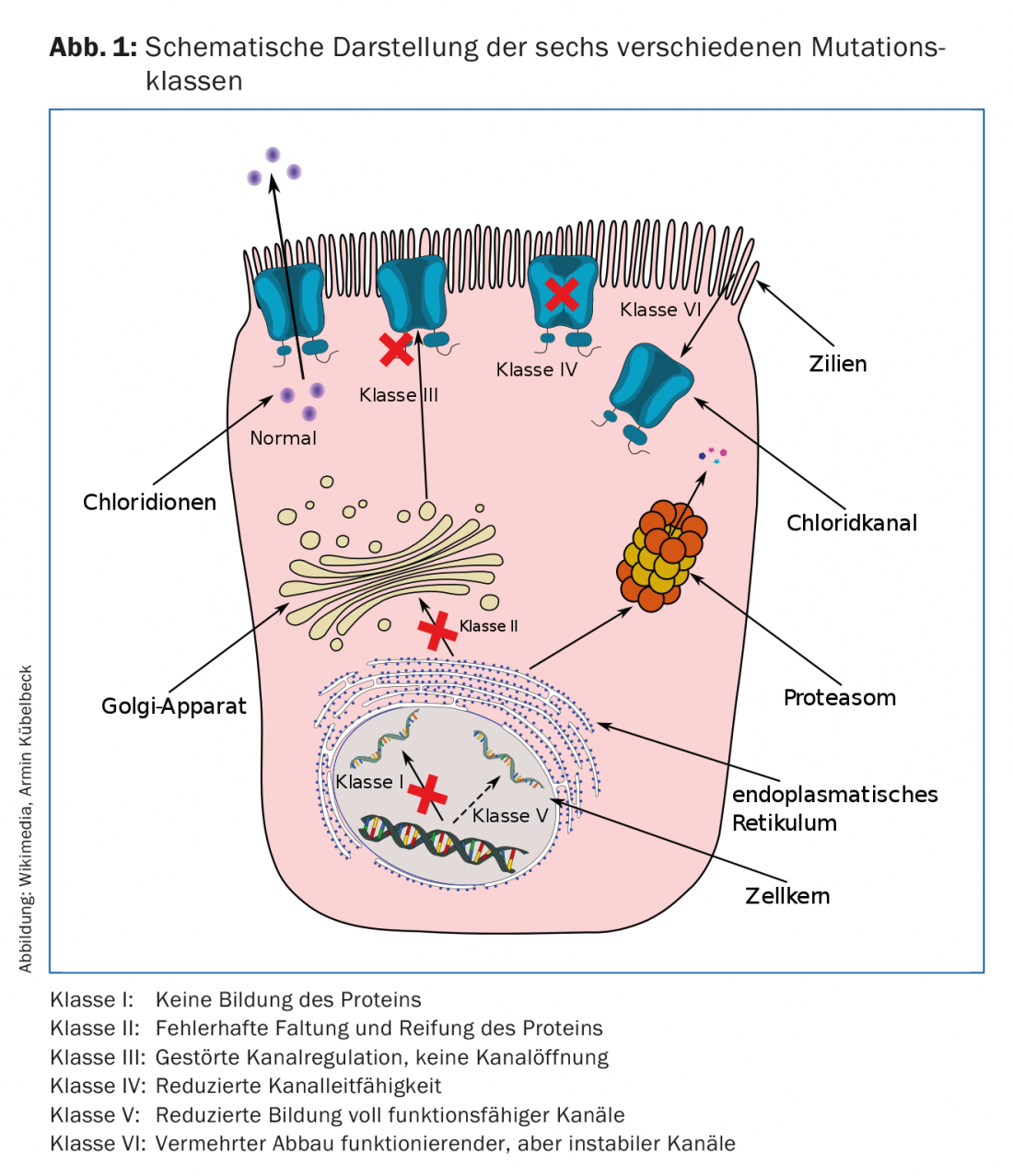
Il gene CFTR sul cromosoma 7 codifica per il canale del cloruro nella membrana cellulare. Le mutazioni genetiche, di cui oggi si conoscono più di 2000 ma meno di 200 sono chiaramente causa di malattia, causano un malfunzionamento o la completa assenza del canale del cloruro nelle cellule epiteliali. Le mutazioni del gene CFTR sono suddivise in sei classi (Fig. 1), che si differenziano per il loro pato-meccanismo. Alcune mutazioni, ad esempio, causano un fallimento quasi completo della sintesi della proteina CFTR. La mutazione 3905insT, che si verifica negli svizzeri o nelle persone di origine svizzera (ad esempio gli Amish in Nord America), è assegnata a questa classe I ed è la seconda mutazione più frequente in Svizzera. In altre mutazioni, l’incorporazione della proteina nella membrana cellulare è impedita o il canale ionico della proteina è bloccato o ha una conduttività limitata. La mutazione più frequente in Europa, ma anche in Svizzera (86% eterozigoti, 47% omozigoti) è assegnata alla classe II e riguarda una delezione in posizione 508 (F508del). La proteina prodotta viene ripiegata in modo errato e degradata prima che venga assunta una funzione. Il fallimento del canale del cloruro porta all’interruzione del trasporto transepiteliale in tutti gli organi in cui le cellule epiteliali esprimono CFTR. Prendendo come esempio l’epitelio delle vie aeree, si verifica una cascata infiammatoria. L’aumento della viscosità del muco causa un’alterazione della clearance mucociliare, che a sua volta è associata a una colonizzazione batterica precoce e, nel prosieguo, cronica. La risposta immunitaria dell’organismo causa un reclutamento eccessivo di granulociti neutrofili, un rilascio inadeguato di elastasi neutrofila e infine la distruzione dei tessuti. Oltre alle due mutazioni classiche elencate come esempio (F508del, 3905insT), che sono responsabili di un decorso grave della malattia, sono ora note anche mutazioni associate a un quadro clinico piuttosto lieve – mentre nel caso di altre mutazioni non è chiaro se siano addirittura causative della FC. Il database www.cftr2.org cerca di fornire una panoramica continuamente aggiornata delle mutazioni note che causano la FC e dei loro probabili fenotipi [1,3,4].
Diagnostica
Prima del 2011, a circa il 10% dei bambini con FC veniva diagnosticato l’ileo da meconio, ma la stragrande maggioranza veniva identificata solo durante il decorso della malattia, quando comparivano i sintomi sospetti della FC. Il gold standard per la diagnosi è il test del sudore, cioè la determinazione quantitativa della concentrazione di cloruro nel sudore dopo la ionoforesi di pilocarpina. Inoltre, la misurazione della conduttività del sudore viene spesso utilizzata come test di screening, in quanto questo test funziona in modo rapido e affidabile con una quantità di sudore significativamente inferiore. Macroduct® e Nanoduct® sono sistemi di test disponibili in commercio. Ai fini della garanzia di qualità, l’esecuzione dei test di saldatura deve essere riservata a centri appropriati.
Nel 2011, la FC è stata inclusa nel programma di screening neonatale della Svizzera. Il tripsinogeno immunoreattivo (IRT) viene misurato nel sangue del tallone. Se il limite viene superato, i neonati vengono indirizzati a un centro pediatrico per la FC. Lì, la diagnosi viene confermata o esclusa con un test del sudore e, se necessario, con ulteriori esami (determinazione dell’elastasi pancreatica, analisi genetica).
Nel 2016, 143 bambini sono stati diagnosticati con la FC attraverso lo screening neonatale. Su un totale stimato di 900 pazienti affetti da FC, circa il 15% di tutti i pazienti potrebbe già essere individuato con l’aiuto dello screening neonatale. Questo valore continuerà a crescere costantemente.
Per il medico generico in particolare, tuttavia, è importante notare che, a causa dei diversi fenotipi, ci possono essere casi diagnosticati nell’infanzia, così come quelli con un decorso lieve o con CFTR residua misurabile. I soggetti possono quindi manifestarsi solo in età adulta con sintomi atipici come pancreatite, sinusite, polipi nasali, bronchiectasie diffuse e/o un desiderio insoddisfatto di avere figli. Pertanto, il medico di base dovrebbe considerare la FC come una possibile diagnosi differenziale in caso di sintomi corrispondenti [1,5–7].
Terapia e assistenza
Secondo le linee guida internazionali, i pazienti dovrebbero essere collegati a centri specializzati. I pazienti vengono visitati a intervalli di 3 mesi. A seconda del decorso individuale della malattia, la terapia deve essere rivista e adattata. Oltre all’anamnesi e all’esame clinico, le misurazioni della funzionalità polmonare (a seconda dell’infrastruttura, pletismografia corporea, spirometria, washout a respiro multiplo N2) e i test microbiologici (espettorato o tampone della gola) fanno parte degli esami di routine, che vengono integrati a intervalli più ampi da imaging dei polmoni e dell’addome, esami del sangue, carichi di glucosio e misurazioni della densità ossea. L’obiettivo è rilevare i cambiamenti in una fase precoce, in modo da poterli contrastare e prevenire un ulteriore deterioramento.
Il trattamento di base è complesso e comprende la bonifica intensiva delle vie aeree mediante inalazione e una speciale fisioterapia respiratoria (soluzione fisiologica ipertonica, rhDNAse se necessario, speciale “tecnica di bonifica delle vie aeree”), la sostituzione dell’enzima pancreatico (lipasi) e la terapia nutrizionale (sostituzione delle vitamine liposolubili, dieta ipercalorica), nonché un trattamento antimicrobico aggressivo (inalazione e/o applicazione sistemica).
A seconda dell’età del paziente e delle circostanze che lo accompagnano, è necessaria anche una consulenza continua e mirata, per poter accettare in modo ottimale le sfide della malattia e i suoi effetti sullo stile di vita. Oltre agli ovvi contenuti legati alla malattia e alla terapia, i pazienti (e, a seconda dell’età, anche i loro tutori legali) hanno bisogno di un supporto per affrontare le questioni sociali, psicologiche, finanziarie e assicurative (invalidità e/o assicurazione sanitaria), per organizzare l’assistenza ai bambini, la frequenza di asili e scuole, la formazione e il lavoro, i viaggi, il desiderio dei genitori di avere di nuovo figli o la pianificazione familiare del paziente adulto stesso, ecc.
Bisogna anche tenere conto del fatto che le persone colpite stanno fortunatamente invecchiando sempre di più. Improvvisamente i pazienti, ma anche i team curanti, sono esposti a problemi interni che sorgono indipendentemente dalla FC, ma ne influenzano il decorso. Per soddisfare queste esigenze, è necessario un approccio terapeutico multidisciplinare, che può essere fornito solo da un centro FC [8,9].
Prospettiva
Nel 2014, Swissmedic ha approvato il farmaco ivacaftor (Kalydeco®) per i pazienti con la mutazione di classe III G551D. Per la prima volta, una terapia causale con un aumento significativo della funzione del canale del cloruro, misurabile in un netto miglioramento del test del sudore e della funzione polmonare, poteva essere utilizzata nei pochi pazienti con questa mutazione (circa il 4-5% di tutti i pazienti in tutto il mondo). Altri modulatori CFTR sono attualmente in fase di sperimentazione. La speranza rimane quella che una terapia causale specifica per la mutazione sia disponibile per un gruppo più ampio di pazienti in futuro [10].
Messaggi da portare a casa
- Grazie ai miglioramenti nella terapia sintomatica e nella gestione della malattia, la qualità della vita e l’aspettativa di vita sono in continuo aumento.
- Già oggi, la metà dei pazienti svizzeri affetti da FC è in età adulta.
- Lo screening neonatale per la FC introdotto nel 2011 consente una diagnosi precoce. Ciò significa che la terapia di base può essere iniziata prima che si verifichino i primi cambiamenti legati alla malattia.
- Sono possibili decorsi oligosintomatici e atipici. Pertanto, la FC deve essere considerata come una possibile diagnosi differenziale in età adulta.
- I chiarimenti e le ulteriori cure devono sempre avvenire in collaborazione con un centro FC.
Letteratura:
- Elborn JS: Fibrosi cistica. Lancet 2016 Nov 19; 388(10059): 2519-2531.
- Zolin A, et al: Relazione annuale ECFSPR 2014. 2016.
- Hergersberg M, et al.: Una nuova mutazione, 3905insT, rappresenta il 4,8% di 1173 cromosomi FC in Svizzera e causa un fenotipo grave. Hum Genet 1997 Aug; 100(2): 220-223.
- CFTR2. www.cftr2.org
- Torresani T, et al: Screening neonatale per la fibrosi cistica in Svizzera – conseguenze dopo l’analisi di uno studio pilota di 4 mesi. J Cyst Fibros 2013; 12(6): 667-674.
- Screening neonatale Svizzera. www.neoscreening.ch
- Barben J, et al: Screening neonatale per la fibrosi cistica – una storia di successo. Schweiz Med Forum 2013; 13(49): 1010-1012.
- Smyth AR, et al: Standard di cura della Società Europea di Fibrosi Cistica: linee guida di miglior pratica. J Cyst Fibros 2014 maggio; 13(Suppl 1): S23-42.
- Elborn JS, et al: Rapporto della task force della European Respiratory Society/European Cystic Fibrosis Society sulla cura degli adulti con fibrosi cistica. Eur Respir J 2016 Feb; 47(2): 420-428.
- Ramsey BW, et al: Un potenziatore CFTR nei pazienti con fibrosi cistica e la mutazione G551D. N Engl J Med 2011 Nov 3; 365(18): 1663-1672.
PRATICA GP 2017; 12(11): 31-33









