Grâce à l’introduction de nouveaux médicaments, le pronostic des patients atteints de myélome multiple n’a cessé de s’améliorer au cours des dernières années. Les diagnostics ont également été élargis, notamment grâce aux analyses cytogénétiques qui permettent une stratification plus précise des risques. Néanmoins, le taux de survie à 5 ans au stade III n’est que de 40%, il y a donc encore de la place pour l’innovation.
En tant que lymphome à cellules B avec prolifération monoclonale de plasmocytes dans la moelle osseuse, le myélome multiple (MM) est une maladie extrêmement hétérogène. Alors qu’environ un quart des personnes atteintes sont asymptomatiques au moment du diagnostic, il existe également des évolutions aiguës avec une destruction osseuse rapide, un dysfonctionnement rénal prononcé, une fatigue, une hypercalcémie et une tendance aux infections [1]. Le nombre de nouveaux cas augmente avec le vieillissement de la population, car la plupart des cas de maladie surviennent entre 70 et 80 ans. Le myélome multiple reste une maladie rare, avec une incidence d’environ 6 cas pour 100 000 habitants chez les hommes et 4 cas pour 100 000 habitants chez les femmes, mais on peut s’attendre à une augmentation d’un tiers du nombre de cas d’ici 2040, rien qu’en raison du changement des structures d’âge [2,3]. Les exigences d’une gestion efficace avec des options thérapeutiques efficaces et des diagnostics ciblés vont donc continuer à augmenter à l’avenir.
Regard sur la physiopathologie
Dans le myélome multiple, les plasmocytes malins produisent des immunoglobulines monoclonales complètes ou incomplètes. Celles-ci peuvent être détectées dans le sérum et l’urine sous forme de chaînes légères à multiplication clonale et portent également le nom de “paraprotéines”. Dans l’électrophorèse des protéines sériques, ils apparaissent sous la forme du fameux “gradient M”. La concentration élevée de ces immunoglobulines peut provoquer différents symptômes, comme l’amylose AL due au dépôt de protéines mal repliées. En outre, un syndrome d’hyperviscosité peut survenir et, beaucoup plus fréquemment, une détérioration de la fonction rénale. En effet, les chaînes légères formées en plus grande quantité sont filtrées au niveau glomérulaire et ne peuvent souvent pas être complètement absorbées dans les tubules. Cela se traduit par une excrétion urinaire appelée “protéinurie de Bence-Jones”. En outre, les paraprotéines s’accumulent dans les glomérules et précipitent dans le tubule distal en se liant à la protéine Tamm-Horsfall produite par les cellules épithéliales tubulaires. Les résultats : Glomérulosclérose et néphropathie de Cast [4].
Les autres symptômes du myélome multiple sont principalement dus au déplacement de l’hématopoïèse normale et à la destruction des os. Ces mécanismes entraînent notamment des douleurs osseuses, des fractures pathologiques, une hypercalcémie, une anémie et une prédisposition aux infections [1]. Souvent, les plaintes des personnes concernées ne sont pas spécifiques, de sorte qu’il n’est pas rare que plusieurs mois s’écoulent avant que le diagnostic correct ne soit posé.
La génétique du myélome multiple est aussi variée que son aspect clinique. Des trisomies sont détectées chez environ 40% des patients. Les translocations impliquant le locus de la chaîne lourde des immunoglobulines (IgH) sur le chromosome 14q32 sont également fréquentes. Elles font partie, avec les trisomies, des altérations génétiques primaires et peuvent être détectées dès les stades préliminaires de la maladie [5]. Au fur et à mesure de l’évolution de la maladie, des aberrations génétiques secondaires, telles que les mutations del(1p) ou RAS, viennent s’ajouter et conditionnent la clinique, la réponse au traitement et le pronostic [3,6].
En fin de compte, l’étiologie du myélome multiple reste inexpliquée. Les cas familiaux sont rares et les facteurs de risque restent à ce jour inconnus [3]. La maladie se développe généralement à partir d’une gammapathie monoclonale de signification indéterminée (MGUS) [7]. En présence de ce précurseur, le risque de progression vers un myélome multiple ou une autre maladie lymphoproliférative nécessitant un traitement est d’environ 1% par an [8]. Cependant, le dépistage précoce des stades précancéreux n’est pas établi, car il n’a pas encore permis d’améliorer significativement le pronostic [3].
Le diagnostic en mutation
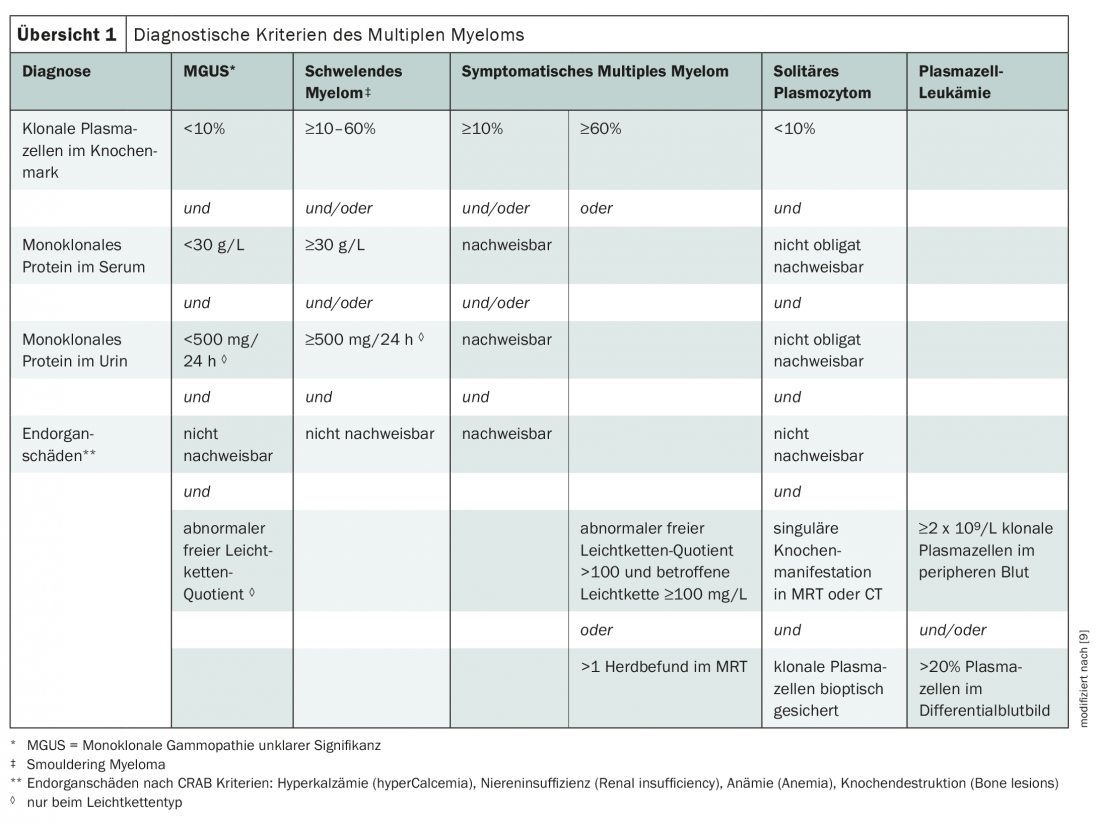
Pour le diagnostic du myélome multiple, on utilise actuellement en premier lieu les critères de l’International Myeloma Working Group (aperçu 1) [3,9]. La maladie est classée selon le type de paraprotéine, les myélomes IgG et IgA étant les plus fréquents. Lorsque seules des immunoglobulines incomplètes, c’est-à-dire des chaînes légères, sont produites, on parle de “myélome à chaînes légères”. Ils représentent environ un cinquième des cas [3].
En plus de l’anamnèse, de l’examen physique, de l’hémogramme et du laboratoire, un scanner corps entier à faible dose et une ponction de moelle osseuse font partie du diagnostic initial. L’imagerie permet de détecter l’ostéolyse et l’ostéopénie [3]. Le cas échéant, elle peut être complétée par un examen IRM pour différencier plus précisément les foyers osseux et diagnostiquer les manifestations extramédullaires. Cela peut notamment être utile pour faire la distinction avec le myélome qui couve [10]. Les examens FDG-PET peuvent également fournir des informations sur les foyers extramédullaires et la réponse thérapeutique, mais ne font actuellement pas partie des examens standard [11]. Au plus tard avant l’introduction d’un traitement, une IRM de régions spécifiques doit être demandée en cas de suspicion d’atteinte extramédullaire et de symptômes neurologiques. Une échocardiographie est également indiquée en cas de suspicion d’amylose cardiaque [3].
Le diagnostic génétique joue un rôle de plus en plus important dans la prise en charge du myélome multiple. Les analyses sur del(17p), t(4 ; 14) et t(14 ; 16) correspondent aujourd’hui au work-up minimal [3]. Ces modifications génétiques s’accompagnent d’un pronostic nettement moins favorable. D’autres aberrations, ainsi que des scores pronostiques basés sur l’expression des gènes, sont certes importants pour le pronostic, mais ne sont pas (encore) prédictifs de traitements spécifiques [6,12].
Classification pour l’évaluation du pronostic
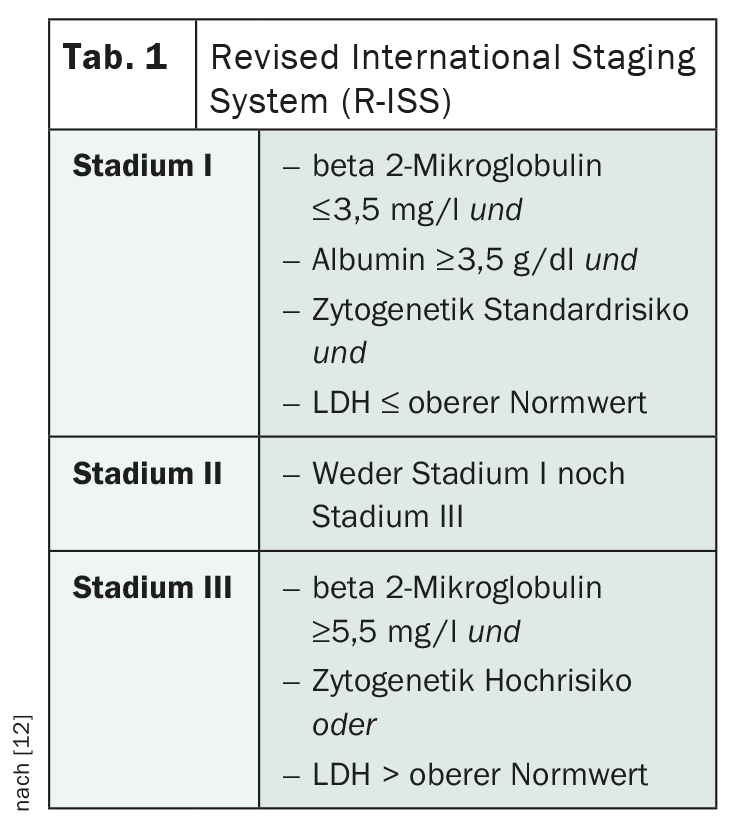
La stadification du myélome multiple joue un rôle important dans l’estimation du pronostic et l’évaluation du traitement le plus approprié. Dans ce contexte, la classification de Salmon et Durie, utilisée depuis longtemps, est aujourd’hui dépassée. Le système international de stadification révisé (R-ISS) de l’International Myeloma Working Group (IMWG) est utilisé à la place (tableau 1) [12]. Cette stadification permet de classer les personnes atteintes en trois groupes pronostiques en tenant compte de l’albumine sérique, de la bêta 2-microglobuline, de la LDH et des aberrations cytogénétiques.
La présence d’une maladie résiduelle minimale (MRD) après traitement est également importante en termes de pronostic. Elle est présente chez la grande majorité des patients même après l’obtention d’une rémission complète selon les critères actuels et est corrélée à un moins bon résultat [13]. Les analyses de MRD n’ayant jusqu’à présent aucune valeur prédictive pour la suite du traitement, elles ne constituent actuellement pas un standard dans les examens de suivi [3].
Aperçu du traitement de première ligne
Outre les évolutions symptomatiques et les critères CRAB (hypercalcémie – C, insuffisance rénale – R, anémie – A et atteinte osseuse – B), il existe également d’autres paramètres radiologiques et sérologiques qui, selon les critères de l’IMWG, constituent des indications pour l’introduction d’un traitement. Il s’agit notamment de la détection de biomarqueurs définissant le myélome, d’un taux de plasmocytes clonaux dans la moelle osseuse de >60%, d’un quotient de chaînes légères libres dans le sérum de >100 (chaînes légères affectées/non affectées) et de lésions focales de plus de 1 cm à l’IRM [3].
Lorsqu’il existe une indication de traitement, on distingue en premier lieu les patients qui peuvent bénéficier d’une transplantation de cellules souches de ceux qui doivent être traités sans thérapie à haute dose en raison de comorbidités. Si la transplantation n’est pas envisageable, différentes combinaisons de deux ou trois médicaments peuvent être utilisées dans le traitement médicamenteux. Les patients souffrant d’insuffisance rénale, d’une activité élevée de la maladie et d’une cytogénétique défavorable devraient de préférence recevoir un traitement avec le bortézomib comme composant [3]. Les autres agents utilisés sont le cyclophosphamide, le melphalan, le lénalidomide et les stéroïdes. Malheureusement, seuls quelques-uns des schémas les plus courants ont été directement comparés entre eux. Dans l’ensemble, les combinaisons de trois substances utilisant un inhibiteur du protéasome, un immunomodulateur et un stéroïde semblent être supérieures aux combinaisons de deux substances. Si l’état général est bon, elles sont donc préférables [3].
S’il existe une possibilité d’autogreffe de cellules souches, celle-ci constitue le traitement de choix en première ligne. Jusqu’à présent, aucun nouveau médicament n’a pu rivaliser avec la transplantation en termes de taux de rémission, de profondeur de réponse et de survie sans progression [14,15]. Les effets indésirables de la thérapie à haute dose constituent une limite à l’indication d’une autogreffe de cellules souches. Celle-ci suppose un bon fonctionnement des organes et l’absence de comorbidités significatives [16]. Il y a quelques années encore, on recommandait un traitement d’induction contenant de la vincristine et de l’anthracycline. Elle a depuis été remplacée par des thérapies combinées avec les nouvelles substances actives thalidomide, bortezomib et lénalidomide, qui entraînent des taux de réponse nettement meilleurs. Là encore, les données de comparaison directe entre les différentes combinaisons thérapeutiques sont malheureusement limitées, des rémissions complètes étant obtenues dans 20 à 40% des cas [3]. Actuellement, le choix du traitement d’induction est principalement guidé par les facteurs individuels du patient, les effets secondaires et la disponibilité des médicaments.
La transplantation de cellules souches qui suit le traitement d’induction peut se faire en solo ou en tandem. Dans ce dernier cas, une seconde autogreffe est réalisée dans les six mois. Chez les patients en stade III du R-ISS, on a observé non seulement une survie sans progression plus longue, mais aussi une survie globale plus longue [17]. Cependant, la toxicité accrue d’une deuxième thérapie à haute dose doit également être prise en compte. Actuellement, la transplantation en tandem est recommandée pour les patients du groupe III du R-ISS et pour les personnes présentant une cytogénétique à haut risque, en tenant compte de différentes analyses de sous-groupe et à long terme [3]. Il faut bien entendu s’assurer de la présence d’un nombre suffisant de cellules souches autologues conservées. Pour le traitement à haute dose, on utilise aujourd’hui le melphalan 200 mg/m2 [18]. Les régimes de conditionnement alternatifs utilisant le cyclophosphamide et/ou l’irradiation corporelle totale ne sont plus recommandés en raison de leur toxicité plus élevée. Le traitement optimal à haute dose fait également l’objet de plusieurs études à l’heure actuelle, et l’ajout de busulfan n’a pas permis de prolonger la survie globale. L’ajout de bortézomib ne s’est malheureusement pas non plus révélé efficace [19]. Il n’existe pas de données concluantes sur la mise en œuvre d’un traitement de consolidation après une autogreffe de cellules souches. Ce traitement peut être particulièrement utile chez les patients présentant une réponse sous-optimale après la transplantation ou en cas de cytogénétique à haut risque [20].
Les recommandations concernant le traitement d’entretien sont plus claires. Après avoir été déconseillée pendant des années, elle fait aujourd’hui partie intégrante des normes thérapeutiques, grâce aux nouveaux médicaments. Ainsi, le bortézomib ou le lénalidomide sont utilisés dans le groupe à haut risque et le lénalidomide dans le groupe à risque standard. Et ce, même en cas de réponse complète [3,19,21].
Un facteur thérapeutique important à ne pas oublier, quel que soit le statut de la transplantation, est l’ostéoprotection. En particulier en cas d’atteinte osseuse et pendant un traitement par glucocorticoïdes, il convient d’utiliser des bisphosphonates ou, surtout en cas d’insuffisance rénale, le dénosumab, afin de prévenir au mieux la résorption osseuse. Le zolendronate est le bisphosphonate de premier choix dans le myélome multiple [3].
Et en cas de progression ou de récidive ?
Les progressions et les récidives après un traitement de première ligne représentent un défi majeur dans le myélome multiple. Notamment parce que le collectif de patients est encore plus inhomogène avec des lignes de traitement ultérieures. Les personnes concernées ont reçu différents traitements et ont vécu différentes expériences. Le choix des médicaments dépend notamment de l’efficacité et de la tolérance de la première ligne. Ainsi, si l’expérience est concluante en première ligne, un médicament de la même classe de substances peut être utilisé en deuxième ligne. En revanche, en cas de faible efficacité ou de mauvaise tolérance, il est indiqué de changer de classe de médicaments. Il existe actuellement une pléthore de nouveaux médicaments et de combinaisons de substances actives. Il est donc possible de choisir parmi un large éventail de substances et de séquences en fonction de l’aspect clinique, des traitements antérieurs et des comorbidités [3].
Alors que les récidives précoces et tardives sont de préférence traitées par transplantation de cellules souches, il existe divers régimes thérapeutiques pour toutes les autres récidives et en cas de contre-indications à la transplantation. Ils sont soit basés sur un inhibiteur du protéasome, soit sur un immunomodulateur et se composent de deux à trois substances actives (aperçu 2). Le daratumumab, un anticorps anti-CD38, est également utilisé ici. En règle générale, les combinaisons triples sont plus efficaces que les combinaisons doubles, mais il faut aussi tenir compte d’un plus grand nombre de toxicités [3,19].

La recherche croissante de nouvelles molécules pour le traitement du myélome multiple a permis d’obtenir quelques succès ces dernières années, en particulier dans le traitement d’entretien et dans les lignes de traitement avancées. Cependant, le pronostic dans le groupe à haut risque et en cas de maladie récidivante est encore défavorable aujourd’hui. L’analyse cytogénétique, de plus en plus répandue, permet déjà de mieux stratifier les risques. Celle-ci suscite l’espoir de disposer à l’avenir non seulement d’options diagnostiques, mais aussi potentiellement d’options thérapeutiques plus ciblées.
Littérature :
- Kyle RA, et al : Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003 ; 78(1) : 21-33.
- NICER : National statistics on cancer incidence. www.nicer.org/en/statistics-atlas/cancer-incidence (dernier accès le 27.02.2021)
- Wörmann B, et al : Myélome multiple. Ligne directrice Onkopedia. www.onkopedia.com/de/onkopedia/guidelines/multiples-myelom/@@guideline/html/index.html (dernier accès le 27.02.2021)
- Dimopoulos MA, et al : Pathogenesis and treatment of renal failure in multiple myeloma. Leukemia (Leucémie). 2008 ; 22(8) : 1485-1493.
- Mikulasova A, et al. : Le spectre des mutations somatiques dans la gammapathie monoclonale de signification indéterminée indique un paysage génomique moins complexe que celui du myélome multiple. Haematologica. 2017 ; 102(9) : 1617-1625.
- Bergsagel PL, Chesi MV : Classification moléculaire et stratification du risque de myélome. Hematol Oncol. 2013 ; 31 Suppl 1(0 1) : 38-41.
- Landgren O, et al. : La gammopathie monoclonale de signification indéterminée (MGUS) précède généralement les myélomes multiples : une étude prospective. Le sang. 2009 ; 113(22) : 5412-5417.
- Dispenzieri A, et al : Prévalence et risque de progression de la gammapathie monoclonale à chaînes légères de signification indéterminée : une étude de cohorte rétrospective basée sur la population. Lancet . 2010 ; 375(9727) : 1721-1728.
- Rajkumar SV, et al : International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014 ; 15(12) : e538-548.
- Dimopoulos MA, et al : Role of magnetic resonance imaging in the management of patients with multiple myeloma : a consensus statement. J Clin Oncol. 2015 ; 33(6) : 657-664.
- Cavo M, et al : Role of (18)F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders : a consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017 ; 18(4) : e206-e17.
- Palumbo A, et al : Système international de stadification révisé pour le myélome multiple : un rapport du groupe de travail international sur le myélome. J Clin Oncol. 2015 ; 33(26) : 2863-2869.
- Munshi NC, et al : Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma : A Meta-analysis. JAMA Oncol. 2017 ; 3(1) : 28-35.
- Barlogie B, et al : Supériorité de l’autogreffe en tandem sur le traitement standard pour les myélomes multiples précédemment non traités. Le sang. 1997 ; 89(3) : 789-793.
- Gay F, et al : Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma : a randomised, multicentre, phase 3 trial. Lancet Oncol. 2015 ; 16(16) : 1617-1629.
- Merz M, et al : Survival of elderly patients with multiple myeloma-Effect of upfront autologous stem cell transplantation. Eur J Cancer. 2016 ; 62 : 1-8.
- Cavo M, et al : Double Autologous Stem Cell Transplantation Significativement Prolongs Progression-Free Survival and Overall Survival in Comparison with Single Autotransplantation in Newly Diagnosed Multiple Myeloma : An Analysis of Phase 3 EMN02/HO95 Study. Sang . 2017 ; 130 (supplément 1) : 401.
- Giralt S : 200 mg/m(2) melphalan–the gold standard for multiple myeloma. Nat Rev Clin Oncol. 2010 ; 7 : 490-491.
- Dimopoulos MA, et al : Multiple myeloma : EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annales d’Oncologie. 2021 ; 32(3) : 309-322.
- Einsele H, et al : Response-adapted consolidation with bortezomib after ASCT improves progression-free survival in newly diagnosed multiple myeloma. Leukemia . 2017 ; 31(6) : 1463-1466.
- McCarthy PL, et al : Lenalidomide Maintenance After Autologous Stem-Cell Transplantation in Newly Diagnosmed Multiple Myeloma : A Meta-Analysis. J Clin Oncol. 2017 ; 35(29) : 3279-3289.
InFo ONKOLOGIE & HÉMATOLOGIE 2021 ; 9(2) : 22-26