Les amyloïdoses sont des maladies de malformation des protéines qui se traduisent généralement par le dépôt extracellulaire de fibrilles protéiques insolubles ayant une configuration en feuillet β antiparallèle. Les mécanismes pathologiques sous-jacents sont multiples. Les causes acquises sont par exemple des concentrations élevées des protéines concernées dans le sang ou les tissus en cas de maladie clonale sous-jacente ou d’inflammation.
Les amyloïdoses sont des maladies de malformation des protéines qui se traduisent généralement par le dépôt extracellulaire de fibrilles protéiques insolubles ayant une configuration en feuillet β antiparallèle [1]. Chez l’homme, on connaît actuellement 36 protéines amyloïdogènes qui peuvent se déposer sous la forme de ces agrégats [2]. Les mécanismes pathologiques sous-jacents sont multiples. Les causes acquises sont par exemple des concentrations élevées des protéines concernées dans le sang ou les tissus en cas de maladie clonale sous-jacente ou d’inflammation. Les amyloïdoses héréditaires, quant à elles, sont généralement dues à des mutations ponctuelles entraînant une modification de la structure tertiaire de la protéine concernée. Les agrégats qui en résultent peuvent être locaux (site de production et de dépôt identique) ou systémiques (sites de production et de dépôt différents) et présentent souvent un tropisme d’organe caractéristique du sous-type d’amylose correspondant. Les formes d’amylose sont nommées par des acronymes selon la nomenclature internationale, la lettre majuscule “A” étant suivie d’une abréviation correspondant à la protéine précurseur concernée [2]. Les amyloïdoses les plus répandues ainsi que leurs principales caractéristiques sont résumées dans le tableau 1.
L’amylose la plus courante dans les pays industrialisés est l’amylose AL systémique. Avec environ 10 cas par million de personnes-années [3], elle fait partie des maladies rares. Elle est généralement causée par des plasmocytes monoclonaux dans la moelle osseuse qui produisent une chaîne légère amyloïdogène. Dans les amyloses localisées, on trouve également des dépôts d’AL-amyloïde dans de nombreux cas. Cependant, contrairement à l’amylose AL systémique, l’amyloïde AL se dépose localement ou de manière multiloculaire et est presque toujours limitée à un système d’organes. Dans ces cas, les cellules plasmatiques ou B clonales sont souvent présentes de manière localisée et une gammapathie monoclonale n’est pas ou très peu détectable dans le sang ou l’urine [4]. On sait encore peu de choses sur la genèse de l’amylose AL locale. Il est probable que dans de nombreux cas, une stimulation locale des cellules B induite par les antigènes joue également un rôle important [5]. En outre, une activité accrue des cellules B dans le cadre d’une maladie rhumatologique inflammatoire sous-jacente peut en être la cause [6,7].
Les symptômes et les options thérapeutiques de chaque amylose sont déterminés par la pathologie sous-jacente et l’atteinte des organes. Le spectre s’étend de l’absence de nécessité de traitement avec une espérance et une qualité de vie intactes à la maladie la plus grave avec un pronostic extrêmement défavorable, en passant par une maladie potentiellement menaçante mais facile à traiter [8,9]. Malheureusement, en raison de la rareté des amyloïdoses et de leurs symptômes souvent non spécifiques, le diagnostic est souvent tardif, à un moment où, dans le cas des amyloïdoses systémiques, les lésions des organes terminaux sont souvent déjà graves et difficilement réversibles. Pour confirmer le diagnostic (sauf dans le cas de l’amylose ATTR du cœur), la détection histologique de matériel typiquement biréfringent en coloration rouge Congo est obligatoire. Le tissu peut être prélevé sur l’organe concerné ou, dans le cas des amyloïdoses systémiques, sur tout autre site de dépôt potentiel. Dans le cas de l’amylose systémique à chaînes légères (AL), l’aspiration de tissu adipeux s’est révélée être une méthode très sensible pour confirmer le diagnostic [10]. Pour le diagnostic différentiel et la classification précise de l’amylose, ainsi que pour le développement d’une stratégie de traitement individuelle, il est recommandé de présenter le patient à un centre d’amylose. Il est notamment tenu compte du sous-type histologique, de la constellation de résultats cliniques ainsi que de l’étendue des lésions organiques et, en conséquence, de la capacité de traitement. La norme pour différencier les différentes formes d’amylose en Allemagne est l’immunohistochimie, qui doit être réalisée par des pathologistes ou des instituts spécialisés [11]. Une nouvelle biopsie n’est généralement pas nécessaire.
Amylose pulmonaire
L’amylose pulmonaire est un diagnostic très rare. La prévalence exacte est toutefois inconnue, car elle est souvent asymptomatique et il s’agit donc souvent de découvertes fortuites, voire post-mortem. D’avril 2000 à octobre 2019, nous avons vu dans notre centre un total de 157 patients atteints d’amylose pulmonaire localisée [6]. Au cours de la même période, nous avons vu 2240 patients atteints de l’amylose systémique la plus courante en Allemagne et en Suisse, l’amylose AL. Des études autopsiques suggèrent que les dépôts amyloïdes pulmonaires et les modifications consécutives du tissu pulmonaire sont détectables dans jusqu’à 90% des cas d’amylose AL systémique [12]. Cependant, la symptomatologie clinique des patients concernés n’est que très rarement attribuée à une atteinte pulmonaire, même en présence d’anomalies morphologiques au scanner [13]. La raison en est que la plupart des patients atteints d’amylose AL systémique ont une atteinte cardiaque cliniquement significative et facile à confirmer par échocardiographie [9]. En revanche, une symptomatologie pulmonaire isolée est plus souvent observée dans les amygdalites AL localisées [6]. Elles représentent également la majorité des amyloses pulmonaires diagnostiquées ante mortem. L’espérance de vie des patients atteints d’amylose AL localisée n’est généralement pas réduite par rapport à la population générale. En outre, le risque de progression vers une amylose AL systémique est généralement faible. Cependant, l’évolution clinique est souvent marquée par des récidives ou des progressions locales.
Selon le site de dépôt de l’amyloïde, on peut distinguer 3 types d’atteinte caractéristiques de l’amylose pulmonaire : trachéobronchique (voies respiratoires supérieures/inférieures), pulmonaire nodulaire et alvéolo-septale diffuse.
Amylose trachéobronchique
Patient 1 : Nous avons vu une patiente âgée de 55 ans au moment du diagnostic, avec un TA légèrement réduit, un IMC de 23 kg/m2, des antécédents de diabète sucré de type 2, d’hypertension artérielle, un abus persistant de nicotine par inhalation (35 paquets-années) et des antécédents de bronchite chronique dans l’enfance. En raison de bronchites récidivantes, des traitements antibiotiques répétés ont été administrés depuis plus d’un an. Une toux chronique était également présente.
A l’extérieur, la radiographie du thorax a montré une atélectasie partielle du lobe moyen ainsi que 2 foyers ronds possibles. Le scanner thoracique avec contraste a également révélé des signes d’atélectasie du lobe moyen avec un épaississement blanchâtre très discret de la bronche du lobe moyen, et encore plus discret de la bronche du lobe supérieur droit. Il y avait également une lymphadénopathie hilaire droite concomitante. Le résultat a été considéré, jusqu’à preuve du contraire, comme une masse avec rétrécissement de la bronche du lobe moyen droit et une métastase ganglionnaire. Une bronchoscopie avec excision d’un échantillon a été réalisée et a révélé des dépôts amyloïdes interstitiels et vasculaires importants de type AL lambda sur le plan histologique. La biopsie de la moelle osseuse qui a suivi n’a pas révélé la présence de plasmocytes ou de cellules B clonaux. Aucune trace d’amyloïde n’a été détectée dans le matériel provenant d’une biopsie du rectum ni dans l’aspiration de tissu adipeux réalisée dans nos locaux. L’échocardiographie n’a pas non plus révélé d’implication cardiaque d’une amylose systémique. Cependant, l’électrophorèse d’immunofixation a révélé une gammapathie monoclonale de type IgG lambda. Nous avons posé le diagnostic suivant : amylose bronchique locale, type AL lambda, avec gammapathie monoclonale concomitante de type IgG lambda.
Après environ un an et demi, une infection exacerbée s’est produite dans le sens d’une progression de l’amylose bronchique avec une occlusion de bronches partielles. Une recanalisation et une dilatation endobronchiques ont été réalisées, ainsi qu’une radiothérapie thoracique au cas par cas à 20 Gy en 10 fractions. Au cours des mois suivants, les symptômes se sont lentement améliorés. Au cours de la période d’observation supplémentaire d’un an dont nous avons connaissance, l’état clinique et radiologique est resté stable. Il n’y avait aucun signe de progression de la gammapathie monoclonale.
Entre avril 2000 et octobre 2019, nous avons vu au Centre de l’amylose de Heidelberg 12 patients atteints d’amylose locale du nasopharynx, 51 patients atteints d’amylose locale du larynx et 31 patients atteints d’amylose locale des voies respiratoires inférieures [6]. Sur le plan physiopathologique, ce type d’atteinte se traduit par l’apparition de plaques amyloïdes multifocales sous-muqueuses sur l’épithélium respiratoire, qui provoquent une obstruction subtotale à totale des voies respiratoires dépendantes. Selon la localisation, on peut distinguer une atteinte proximale, une atteinte moyenne et une atteinte distale [14]. Les patients sont souvent symptomatiques en raison de la toux, des infections respiratoires récurrentes, de l’hémoptysie, du stridor inspiratoire et de l’enrouement. En particulier en cas de sténose proximale de haut degré, par exemple sous-glottique, une dyspnée peut également être induite, qui est souvent attribuée à tort à une origine asthmatique [12].
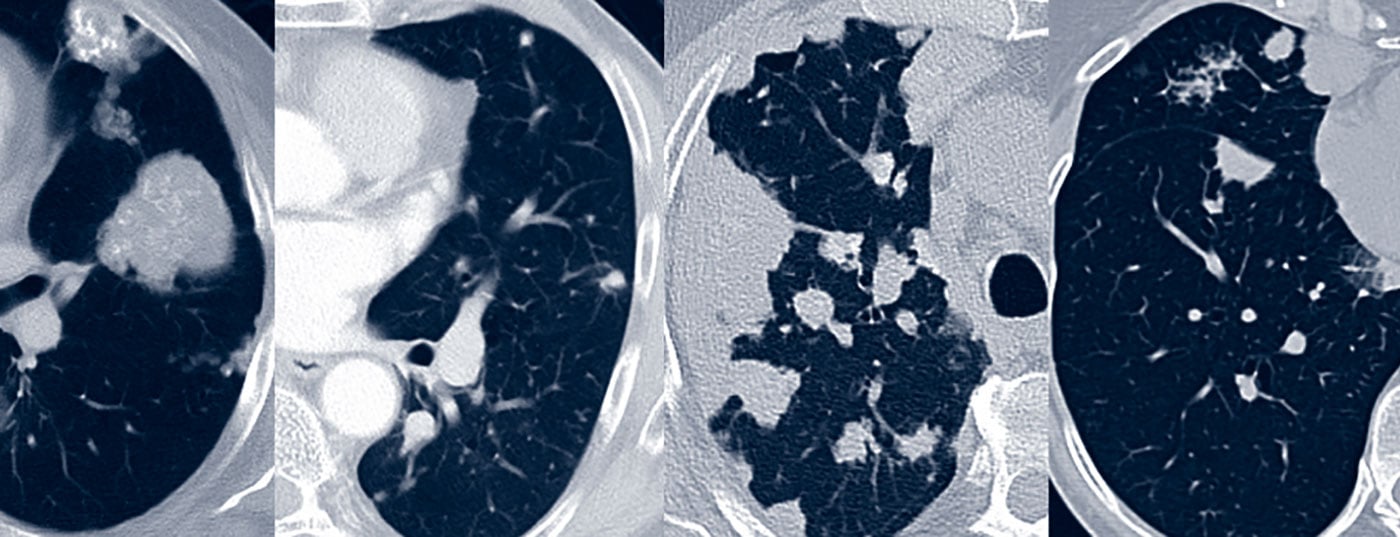
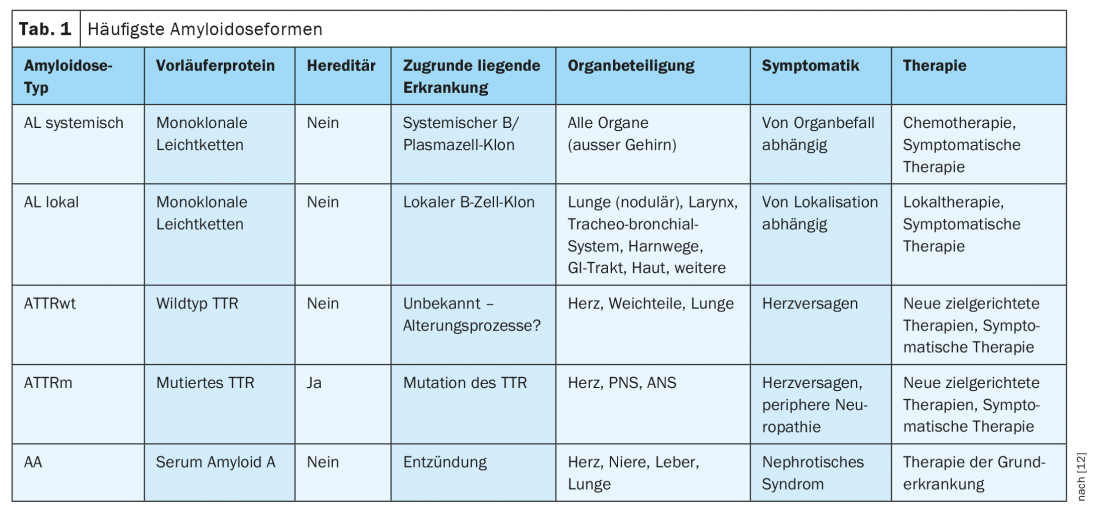
Dans le diagnostic de la fonction pulmonaire, l’obstruction est un élément déterminant de l’amylose trachéobronchique, en particulier en cas d’atteinte proximale [14]. Des épaississements de la paroi de la trachée et/ou des bronches peuvent être détectés par tomodensitométrie (Fig. 1). Les sténoses bronchiques entraînent des dystelectasies lobaires ou segmentaires et peuvent se traduire par des pneumonies de rétention et des destructions du parenchyme pulmonaire. Les calcifications des parois trachéobronchiques sont plus fréquentes. Contrairement à la trachéobronchopathie ostéochondroplastique, la paroi postérieure de la trachée peut également être impliquée [15]. L’examen bronchoscopique révèle la présence de dépôts blanchâtres irréguliers qui peuvent rétrécir la lumière de manière subtotale à totale. Elles sont généralement diffuses et touchent également la paroi postérieure de la trachée. Les lésions sont fragiles et ont tendance à saigner ultérieurement lors des biopsies. Histologiquement, l’amyloïde se trouve dans la sous-muqueuse et les vaisseaux sanguins, souvent associée aux plasmocytes et aux cellules géantes. Dans le sous-type, AL est généralement détectable [16].
Le plus souvent, l’amylose trachéobronchique reste confinée à l’organe et n’est que rarement l’expression d’une amylose AL systémique [16]. Par conséquent, la chimiothérapie systémique n’est généralement pas utile pour cette maladie. En cas d’atteinte importante, par exemple en raison de bronchites récurrentes ou de dyspnée, des mesures thérapeutiques locales sont indiquées. L’ablation endobronchique de l’amyloïde au moyen d’un laser ou de pinces a fait ses preuves. Dans des cas exceptionnels, il est également possible de recourir à une radiothérapie focalisée, qui est potentiellement dirigée contre la maladie clonale à cellules B qui en est la cause. Les infections pulmonaires peuvent mettre la vie en danger à cause des sténoses et nécessitent donc généralement un traitement rapide adapté à l’antibiogramme.
En l’absence d’hémorragies graves ou d’infections pulmonaires, le pronostic de l’amylose trachéobronchique localisée est très bon. Dans moins de 20% des cas, la maladie progresse sur le plan clinique ou morphologique au scanner dans les 5 ans [6].
Amylose nodulaire pulmonaire
Patient 2 : nous avons vu un patient âgé de 49 ans au moment du diagnostic, en bon état général, IMC 28 kg/m². Il n’y avait pas d’antécédents pertinents, à l’exception d’un état d’abus de nicotine par inhalation (15 paquets-années, abstinent depuis 20 ans).
La consultation pneumologique initiale a été motivée par une infection broncho-pulmonaire persistante pendant plusieurs semaines ainsi que par de la fièvre, des sueurs nocturnes et une dyspnée d’effort (poids stable, pas de toux). La fonction pulmonaire était normale et la diffusion n’était pas limitée. Un scanner thoracique avec produit de contraste a été réalisé huit mois avant sa présentation dans notre centre. Plusieurs densités partiellement calcifiées, situées pour la plupart en périphérie, étaient visibles dans les deux poumons et étaient nouvelles ou avaient augmenté en taille par rapport à un examen précédent effectué trois ans auparavant. En outre, plusieurs lésions kystiques ont été observées de part et d’autre, mais aucune lésion spatiale spécifique d’un malignome ni aucun ganglion lymphatique hypertrophié. Le diagnostic différentiel a été posé sur la suspicion de mycobactériose atypique. Une bronchoscopie flexible a été réalisée et a révélé un examen endobronchique normal sans signes tumoraux directs ou indirects. Le lavage broncho-alvéolaire du lobe moyen n’a pas permis de mettre en évidence l’agent pathogène et la mycobactériose n’a pas été détectée.
Cinq mois avant sa présentation chez nous, une ponction guidée par scanner (pneumothorax post-interventionnel sans nécessité de traitement) du lobe inférieur droit a été réalisée. Des dépôts amyloïdes interstitiels AL kappa modifiés de manière régressive ont alors été mis en évidence. La biopsie de la moelle osseuse qui a suivi n’a pas révélé la présence de plasmocytes clonaux ou de cellules B, ni d’amyloïde. Un contrôle de l’évolution par scanner effectué peu avant la présentation dans notre centre a révélé la présence constante de granulomes calcifiés au niveau bipulmonaire.
Lors de sa présentation dans notre centre, nous avons effectué une aspiration du tissu adipeux, qui n’a pas révélé la présence d’amyloïde. L’échocardiographie n’a pas révélé d’implication cardiaque de l’amylose systémique. L’électrophorèse d’immunofixation et la mesure des chaînes légères libres dans le sérum n’ont pas permis d’exclure la présence d’une faible gammapathie monoclonale de type IgG kappa.
Nous avons posé le diagnostic d’amylose pulmonaire locale de type kappa, avec une gammapathie monoclonale concomitante de type IgG kappa. Un suivi à 6 mois n’a montré aucune progression de la gammapathie et n’a toujours pas révélé d’amylose AL systémique. Nous avons recommandé un suivi pneumologique régulier.
Entre avril 2000 et octobre 2019, 63 patients atteints d’amylose nodulaire pulmonaire localisée se sont présentés au Centre de l’amylose de Heidelberg [6]. Sur le plan physiopathologique, cette manifestation se traduit par des amyloïdes tumoraux – des dépôts multifocaux d’amyloïde dans le parenchyme pulmonaire. Dans le sous-type, il s’agit le plus souvent de AL. On pense actuellement que les lymphomes du tissu lymphatique associé à la muqueuse (MALT) sont à l’origine de la plupart des amyloses nodulaires pulmonaires [12]. Il s’agit souvent d’une découverte fortuite à l’imagerie thoracique. Moins de la moitié des patients concernés présentent des symptômes tels que la toux, la dyspnée, l’hémoptysie ou les infections broncho-pulmonaires. Par rapport à l’amylose trachéobronchique, le diagnostic est donc plus tardif et l’âge moyen des patients atteints d’amylose pulmonaire nodulaire est plus élevé que celui des patients atteints d’amylose trachéobronchique (68 ans contre 55 ans).
Le diagnostic de la fonction pulmonaire permet d’espérer une restriction en cas de résultat prononcé. La morphologie tomodensitométrique est généralement dominée par un modèle nodulaire-focal avec des lésions spatiales solitaires à multiples et/ou des foyers ronds. La morphologie des foyers est variée, avec des bords lisses à spiculés, ainsi que des calcifications et une association pleurale facultatives (Fig. 1). La morphologie du scanner ne permet pas de différencier les lésions des affections d’origine maligne ou granulomateuse. Les kystes pulmonaires, parfois avec des foyers ronds associés, et une lymphadénopathie peuvent également faire partie de l’image variée du scanner [17].
Si une néoplasie ou une amylose systémique peuvent être exclues par le diagnostic différentiel, le pronostic de l’amylose ganglionnaire pulmonaire est très bon. En règle générale, la maladie évolue de façon bénigne, et dans moins de 36% des cas, elle progresse sur le plan morphologique par scanner au cours des 5 années suivantes [6]. Les amyloïdes ou les kystes limitant la fonction peuvent être traités par une excision chirurgicale préservant au mieux les tissus environnants.
Amylose alvéolo-septale diffuse
Patient 3 : nous avons vu une patiente âgée de 50 ans au moment du diagnostic, avec un TA réduit et un IMC de 22 kg/m². Cinq mois avant sa première présentation dans notre centre, elle a reçu des traitements antibiotiques et antiobstructifs pour des symptômes de rhume prolongés avec une toux non productive, qui n’ont pas amélioré ses symptômes. En outre, la patiente a signalé une augmentation de l’œdème des chevilles et des jambes depuis environ 6 mois, ainsi que des palpitations et des tachycardies récurrentes. Il y avait une forte réduction de la capacité d’exercice avec dyspnée après avoir monté environ ½ étage ou marché 150 m sur le plat. Jusqu’à il y a quelques semaines, elle a perdu beaucoup de poids (>10% en 6 mois), mais elle en a repris récemment avec des œdèmes cliniquement marqués. L’examen physique a révélé une macroglossie et un gonflement significatif de la base de la langue. Antécédents de morsures de langue récurrentes au cours des mois précédents. De plus, des hémorragies périorbitaires étaient visibles et existaient depuis environ un an. Interrogée, la patiente a indiqué qu’avant le début de la symptomatologie pulmonaire, elle avait bénéficié d’une intervention chirurgicale bilatérale de la main pour un syndrome du canal carpien.
Le scanner thoracique natif a révélé des opacités marquées en verre dépoli bilatérales avec un punctum maximum au-dessus des segments du lobe inférieur. Il y avait également de multiples modifications inflammatoires péribronchovasculaires et surtout sous-pleurales. Il en est résulté une suspicion de lymphome hilaire droit. Une biopsie transbronchique a révélé une amylose alvéolo-septale diffuse de type AL lambda. La biopsie de la moelle osseuse a révélé un taux de plasmocytes de 80% avec une restriction en chaînes légères lambda. Dans le sérum, les chaînes légères libres lambda étaient fortement augmentées à 7572 mg/l (kappa 8 mg/l).
En résumé, nous avons posé le diagnostic d’amylose AL systémique de type lambda. La symptomatologie initiale était notamment pulmonaire et une atteinte pathognomonique des tissus mous (macroglossie, hémorragies périorbitaires, c.-à-d. après une opération du STC bds) a été mise en évidence. Le reste de l’organigramme a révélé une atteinte cardiaque de haut niveau (NT-proBNP 54 749 ng/l, échocardiographie globale montrant une fonction de pompe encore bonne mais nettement diminuée dans le sens longitudinal avec une épaisseur du septum de 18 mm) ainsi qu’une atteinte rénale (protéinurie 8,2 g/d, eGFR 30 ml/min). Nous avons recommandé un traitement systémique relativement bien toléré et efficace à base de bortézomib et de dexaméthasone. Malheureusement, un mois après sa première présentation dans notre centre, il a été victime d’un exitus letalis.
L’infestation alvéolo-septale diffuse s’accompagne d’un tableau clinique de pneumopathie interstitielle. À de rares exceptions près, ces patients présentent une amylose AL systémique comme maladie sous-jacente [12]. L’histopathologie révèle la présence de dépôts amyloïdes dans les parois vasculaires et les septa alvéolaires, qui entravent les échanges gazeux et peuvent entraîner une hypertension artérielle pulmonaire. Le symptôme principal est la dyspnée. La symptomatologie pulmonaire est souvent difficile à évaluer cliniquement dans le contexte d’une maladie cardiaque généralement dominante dans l’amylose systémique.
Le diagnostic de la fonction pulmonaire révèle souvent une restriction et une capacité de diffusion du CO limitée. L’effort entraîne une hypoxémie inadéquate. La tomodensitométrie révèle généralement des augmentations diffuses de la structure pulmonaire, telles que des épaississements septiques, des infiltrats de verre dépoli et des micronodules (figure 1). En outre, on peut observer des lésions spatiales/consolidations, des kystes pulmonaires et une lymphadénopathie [17].
La mise en évidence d’une amylose alvéolo-septale pulmonaire doit inciter à réaliser un bilan complet de la présence d’une amylose AL systémique. Dans de rares cas, le schéma d’atteinte pulmonaire-nodale ou trachéobronchique peut être associé à une amylose systémique [6,17]. S’il y a un mélange des modèles d’infestation, une évolution systémique est même très probable [16]. L’amylose AL systémique devrait donc toujours faire l’objet d’un diagnostic différentiel en cas de suspicion d’amylose pulmonaire locale, en particulier en cas de présence simultanée d’une gammapathie monoclonale.
Si aucune amylose systémique n’est détectée, il est très important de suivre de près l’évolution de la maladie. En présence d’une amylose AL systémique, l’indication d’une chimiothérapie systémique est posée. Le choix du régime de chimiothérapie dépend essentiellement de l’étendue de l’atteinte des organes. Par exemple, la chimiothérapie à haute dose avec autogreffe de cellules souches ne peut plus être envisagée pour les patients présentant une atteinte cardiaque significative ou une capacité de diffusion du CO <50%.
Dans le cadre d’une amylose systémique, les épanchements pleuraux sont fréquents, généralement sous forme de transsudat et dans jusqu’à 1/3 des cas sous forme d’exsudat [12]. Si les épanchements pleuraux dus à une amylose systémique sont réfractaires au traitement diurétique maximal et aux ponctions pleurales, il faut envisager une atteinte pleurale [12].
Diagnostic différentiel de l’amylose pulmonaire locale par rapport à l’amylose AL systémique
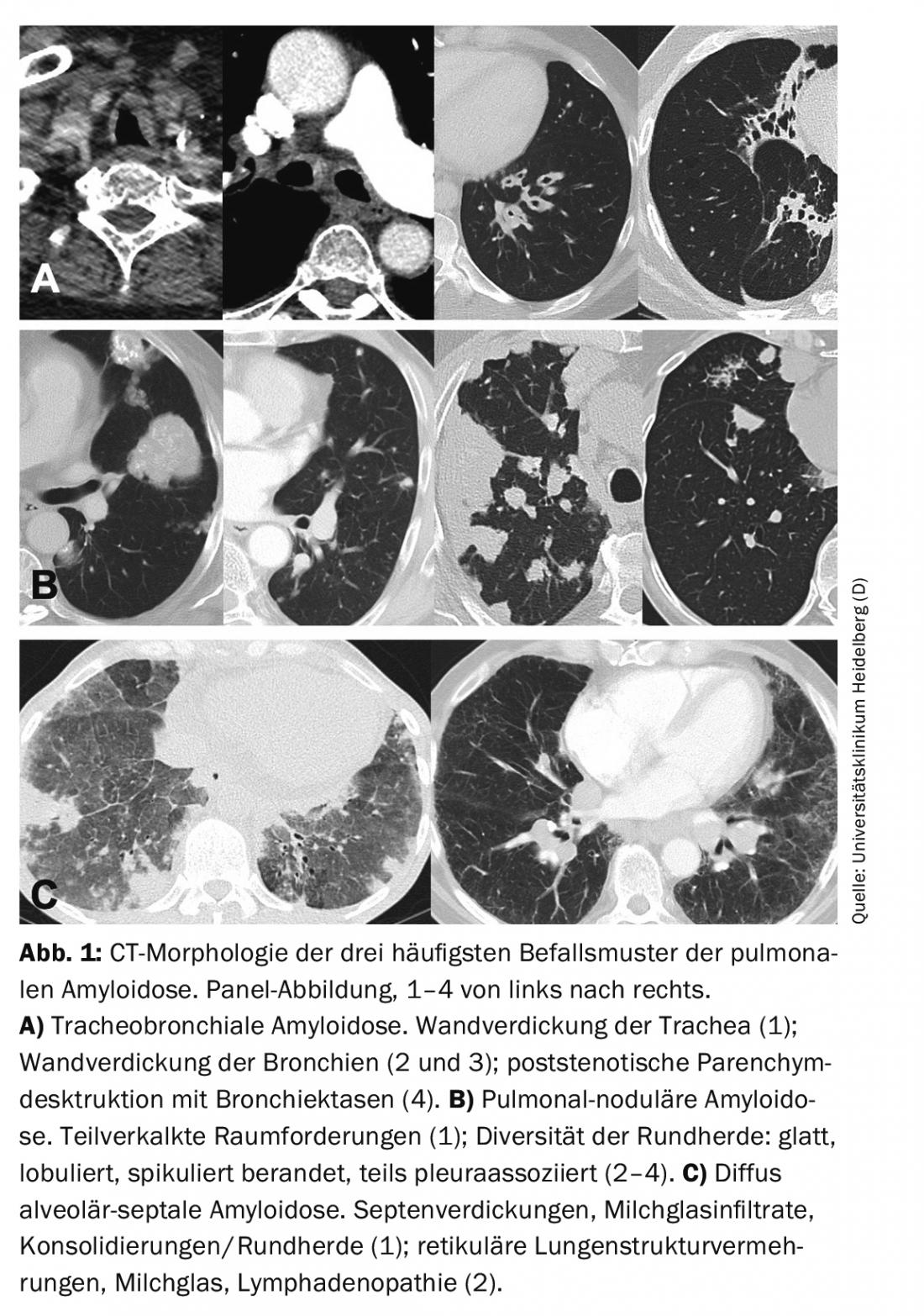
La plupart des amyloïdoses pulmonaires confirmées par l’histologie présentent un sous-type AL. Comme décrit ci-dessus, il s’agit dans la plupart des cas d’amyloïdoses localisées de très bon pronostic. Le diagnostic différentiel entre amylose AL locale et amylose AL systémique est donc particulièrement important, en particulier dans le cas d’une amylose AL pulmonaire nouvellement confirmée (figure 2).
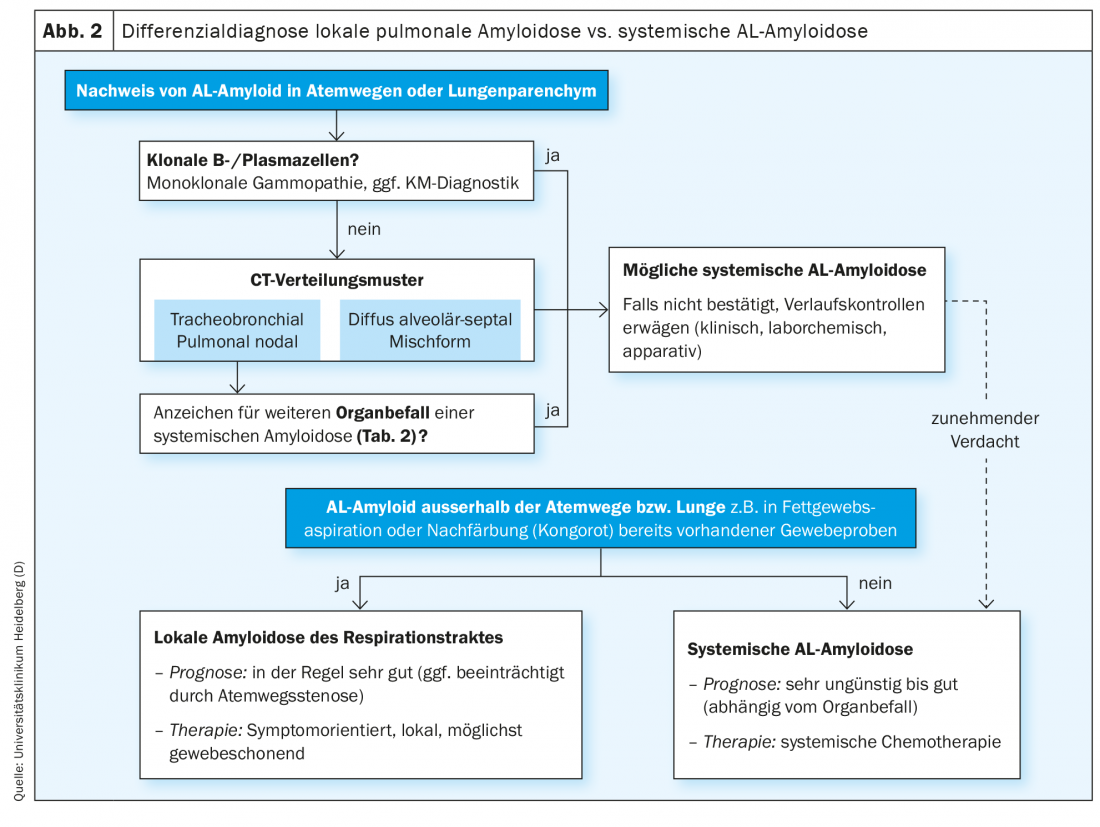
Une anamnèse détaillée et un interrogatoire ciblé sur la symptomatologie concomitante peuvent donner des indications sur un autre système d’organes affecté dans le cadre d’une amylose AL systémique (tableau 2). Les syncopes, l’hypotension artérielle et la dyspnée d’effort inexpliquée au niveau pulmonaire peuvent indiquer une atteinte cardiaque. L’atteinte rénale se manifeste généralement sous la forme d’une atteinte glomérulaire avec syndrome de perte protéique, moins souvent une atteinte réno-vasculaire entraîne directement des troubles de la fonction rénale. La perte constante de protéines peut entraîner une perte de poids en plus d’une urine mousseuse, et la plupart des patients présentent des œdèmes au cours de l’évolution. Une atteinte du tractus gastro-intestinal peut entraîner des diarrhées ou des saignements. Le foie est rarement atteint de manière cliniquement prépondérante. Cependant, des troubles de la coagulation sont souvent observés en raison de la liaison du facteur X de coagulation par l’amyloïde, même dans le cadre d’une atteinte importante des tissus mous. La macroglossie et les hémorragies périorbitaires sont pathognomoniques de l’amylose AL systémique. Un syndrome du canal carpien, en particulier bilatéral, ou l’état après une opération de KTS sont également très suspects, car le rétinaculum flexorum du poignet est un site de dépôt typique et précoce de protéines amyloïdes [18–20]. L’atteinte du système nerveux périphérique peut se traduire, entre autres, par des paresthésies, des troubles de la thermesthésie et un affaiblissement des réflexes propres aux muscles. Une dysrégulation orthostatique importante ainsi qu’une constipation indiquent l’atteinte du système nerveux autonome.
Outre les signes vitaux, l’appareillage complet comprend l’ECG (basse tension périphérique en cas d’atteinte cardiaque, variabilité limitée de la fréquence cardiaque en cas de neuropathie autonome), l’échocardiographie (fonction longitudinale limitée, septum épaissi en cas d’atteinte cardiaque) et l’échographie hépatique. (tableau 2). En outre, les biomarqueurs de laboratoire sont les principaux indicateurs d’une lésion d’organe dans le cadre d’une amylose histologiquement prouvée. (tableau 2).
La recherche d’une éventuelle gammapathie monoclonale est d’une importance capitale pour le diagnostic différentiel entre l’amylose AL systémique et locale. Pour garantir une sensibilité suffisante, il convient de combiner l’électrophorèse d’immunofixation dans le sérum et l’urine et la mesure des chaînes légères libres dans le sérum. Plus d’1/3 des patients atteints d’amylose AL localisée présentent une gammapathie monoclonale généralement faible, mais celle-ci ne correspond au sous-type de dépôts AL (kappa ou lambda) que dans environ la moitié des cas [6]. De plus, environ 22% des patients atteints d’amylose AL pulmonaire locale présentent une maladie auto-immune concomitante, comme le syndrome de Sjögren [6]. Ces observations indiquent que les patients atteints d’amylose AL localisée ont une tendance accrue à former des clones multiples de cellules B, bien que le contexte physiopathologique de cette tendance soit encore inconnu. Une gammapathie monoclonale correspondant au sous-type de dépôts AL peut être l’expression du petit clone local de cellules B sous-jacent à l’amylose locale [4,6,11], mais peut également indiquer la présence d’une amylose AL systémique et doit donc généralement être clarifiée par un diagnostic de la moelle osseuse. Dans le cas d’une amylose AL systémique, des cellules B ou plasmatiques clonales sont généralement détectées dans la moelle osseuse avec une restriction en chaînes légères correspondant au sous-type de dépôts AL. En revanche, si la gammapathie monoclonale ou la population clonale de cellules B/plasma ne correspond pas au sous-type de dépôts AL, il n’y a pas de risque accru d’amylose AL systémique.
Les amyloïdoses pulmonaires sont plus susceptibles d’être associées à l’abus de nicotine par inhalation que l’amylose AL systémique [16,17]. La cause de cette association n’est pas encore claire, mais elle pourrait éventuellement s’expliquer par une augmentation de l’incidence des lymphomes due à des substances toxiques.
Messages Take-Home
- L’amylose pulmonaire est une maladie très rare qui peut être divisée en trois types d’atteinte principaux : trachéobronchique, pulmonaire-nodale et interstitielle ou alvéolo-septale.
- Les patients concernés sont souvent asymptomatiques (en particulier au niveau pulmonaire et nodal) et une atteinte cliniquement symptomatique est généralement bien contrôlée par des mesures locales.
- En cas d’amylose pulmonaire localisée (plus fréquente), le pronostic à long terme est généralement très bon, mais les progressions locales sont fréquentes.
- Cependant, tout schéma d’atteinte de l’amylose pulmonaire peut également être l’expression d’une amylose AL systémique au pronostic potentiellement très défavorable.
- En particulier, la présence d’une gammapathie monoclonale, d’un schéma d’atteinte alvéolo-septale ou d’une symptomatologie évocatrice d’une atteinte systémique doit faire l’objet d’un diagnostic différentiel avec l’amylose AL systémique.
Littérature :
- Merlini G, Bellotti V : Mécanismes moléculaires de l’amyloïdose. N. Engl. J. Med 2003 ; 349(6) : 583-596.
- Benson MD, Buxbaum JN, Eisenberg DS, et al : Amyloid nomenclature 2018 : recommandations du comité de nomenclature de l’International Society of Amyloidosis (ISA). Amyloïde 2018 ; 25(4) : 215-219.
- Kyle RA, Linos A, Beard CM, et al. : Incidence et histoire naturelle de l’amylose systémique primaire dans le comté d’Olmsted, Minnesota, de 1950 à 1989. Blood 1992 ; 79(7) : 1817-1822.
- Stuhlmann-Laeisz C, Schönland SO, Hegenbart U, et al : AL amyloidosis with a localized B cell neoplasia. Virchows Arch 2019 ; 474(3) : 353-363.
- Meijer JM, Schonland SO, Palladini G, et al. : Syndrome de Sjögren et amylose cutanée nodulaire localisée : coïncidence ou entité clinique distincte ? Arthritis Rheum 2008 ; 58(7) : 1992-1999.
- Basset M, Hummedah K, Kimmich C, et al : Localized immunoglobulin light chain amyloidosis : novel insights including prognostic factors for local progression. Am J Hematol 2020 ; 1158-1169.
- Veelken K, Hegenbart U, Schönland SO, Blank N. : Amyloïdose locale et systémique de la chaîne légère chez les patients atteints de maladies rhumatismales. Z Rheumatol 2020 ; 660-668.
- Dittrich T, Benner A, Kimmich C, et al : Performance analysis of AL amyloidosis cardiac biomarker staging systems with special focus on renal failure and atrial arhythmia. Haematologica 2019 ; 104(7) : 1451-1459.
- Dittrich T, Bochtler T, Kimmich C, et al : AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have excellent prognostic. Blood 2017 ; 130(5) : 632-642.
- Kimmich C, Schönland S, Kräker S, et al : Amyloïde dans les smears de la moelle osseuse dans l’amylose systémique à chaînes légères. Amyloïde 2017 ; 24(1) : 52-59.
- Baumgart J-V, Stuhlmann-Laeisz C, Hegenbart U, et al : Local vs. systemic pulmonary amyloidosis-impact on diagnosis and clinical management. Virchows Arch 2018 ; 473(5) : 627-637.
- Milani P, Basset M, Russo F, et al : Le poumon dans l’amyloïdose. Eur Respir Rev 2017 ; 26(145) : 170046.
- Ussavarungsi K, Yi ES, Maleszewski JJ, et al : Clinical relevance of pulmonary amyloidosis : an analysis of 76 autopsy-derived cases. Eur Respir J 2017 ; 49(2) : 1602313.
- O’Regan A, Fenlon HM, Beamis JF, et al : Tracheobronchial amyloidosis. L’expérience de l’Université de Boston de 1984 à 1999. Medicine (Baltimore) 2000 ; 79(2) : 69-79.
- Czeyda-Pommersheim F, Hwang M, Chen SS, et al : Amyloidosis : Modern Cross-sectional Imaging. Radiographics 2015 ; 35(5) : 1381-1392.
- Rech JS, Arnulf B, de Margerie-Mellon C, et al : Amylose des voies respiratoires inférieures : présentation, survie et facteurs pronostiques. Une série de cas consécutifs multicentriques. Am J Hematol 2019 ; 94(11) : 1214-1226.
- Brandelik SC, Heussel CP, Kauczor HU, et al : CT features in amyloidosis of the respiratory system – Comprehensive analysis in a tertiary referral center cohort. Eur J Radiol 2020 ; 129 : 109123.
- Kelly JJ : Neuropathies périphériques associées aux protéines monoclonales : A clinical review. Muscle & Nerve 1985 ; 8(2) : 138-150.
- Haan J, Peters WG : Amyloïde et maladie du système nerveux périphérique. Neurologie clinique et neurochirurgie 1994 ; 96(1) : 1-9.
- Sperry BW, Reyes BA, Ikram A, et al : Amylose ténosynoviale et cardiaque chez les patients subissant une libération du canal carpien. J Am Coll Cardiol 2018 ; 72(17) : 2040-2050.
InFo PNEUMOLOGIE & ALLERGOLOGIE 2020 ; 2(4) : 11-17