L’hépatite auto-immune (AIH) est une maladie chronique et progressive du foie caractérisée par une image histologique d’hépatite d’interface, une hypergammaglobulinémie et la présence d’auto-anticorps circulants. L’AIH survient dans tous les groupes d’âge et touche plus souvent les femmes que les hommes (rapport 3:1). Le spectre de l’HCA s’étend de la maladie asymptomatique à l’insuffisance hépatique, en passant par l’hépatite aiguë ou même fulminante. Les corticostéroïdes sont utilisés pour induire la rémission. En traitement d’entretien, on privilégie l’azathioprine pour réduire le risque de récidive pendant au moins deux ans, de préférence quatre.
L’hépatite auto-immune (AIH) représente 11 à 23% de toutes les hépatopathies chroniques. La fréquence est peut-être sous-estimée, car les hépatites virales sont fréquentes et, en présence d’une hépatite virale chronique, une hépatite auto-immune éventuellement concomitante n’est pas diagnostiquée. L’incidence annuelle chez les Caucasiens est de 0,1 à 1,9/100 000 et la prévalence est de 16,9/100 000 [1]. L’AIH est responsable de 2,6% des transplantations hépatiques en Europe [2] et de 4 à 6% des transplantations hépatiques aux États-Unis [3]. Les femmes sont environ trois fois plus touchées. En principe, la maladie se manifeste dans tous les groupes d’âge, mais dans 50% des cas, elle débute avant l’âge de 30 ans.
Pathogenèse
La pathogenèse exacte de l’AIH reste incertaine. Le tableau clinique reflète une interaction complexe entre une prédisposition génétique, des facteurs déclencheurs, des auto-antigènes et des mécanismes d’immunorégulation, qui aboutit finalement à un processus immunologique (à médiation cellulaire, à médiation par les anticorps ou en combinaison) contre les hépatocytes avec développement d’une inflammation chronique pouvant aller jusqu’à la cirrhose. Les facteurs déclencheurs exacts sont inconnus ; des agents infectieux, médicamenteux et toxiques peuvent être envisagés. Un mimétisme moléculaire entre les antigènes étrangers et les antigènes propres est l’explication la plus courante de la perte de la tolérance au soi [4].
Clinique
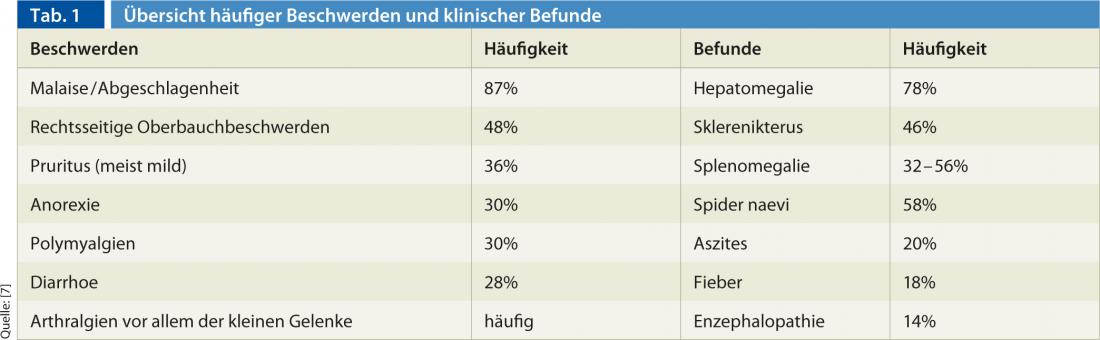
L’AIH a une apparence très hétérogène et fluctuante. Le diagnostic est souvent retardé, car il n’y a souvent que des symptômes légers et non spécifiques d’une hépatite aiguë autolimitée. Le tableau 1 donne un aperçu des symptômes typiques et des résultats cliniques [5].
Il existe différentes formes de manifestation, qui sont brièvement examinées ci-dessous :
- Asymptomatique : le diagnostic est posé sur la base d’une découverte fortuite de valeurs hépatiques élevées chez des patients asymptomatiques.
- Hépatite aiguë : début aigu dans environ 40% des cas. Une anamnèse précise révèle souvent des symptômes non spécifiques antérieurs tels que des épisodes de malaise, des nausées et des arthralgies.
- Insuffisance hépatique aiguë : dans de rares cas, une présentation initiale sous forme d’hépatite fulminante (5%) ou de décompensation hépatique (“burned-out disease”) avec ascite (20%), encéphalopathie hépatique (14%) ainsi que des varices œsophagiennes hémorragiques (8%).
- Cirrhose du foie : jusqu’à 30% des diagnostics initiaux de cirrhose du foie ou de complications associées.Une caractéristique typique de l’AIH est son association à des syndromes extra-hépatiques à médiation immunitaire tels que la thyroïdite auto-immune, le vitiligo, l’alopécie, la colite ulcéreuse, la polyarthrite rhumatoïde, le diabète sucré et la glomérulonéphrite.
Répartition
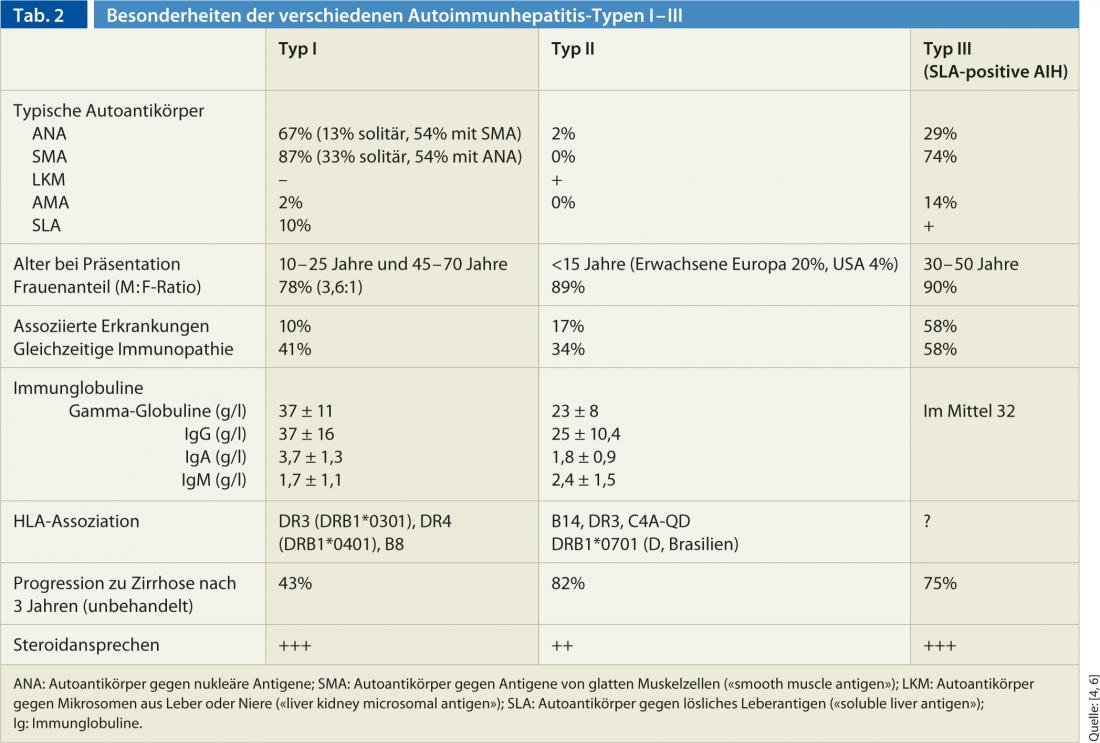
Sur la base de la constellation d’anticorps, l’AIH est classée en trois types différents (type I-III) (tableau 2). L’AIH de type I est la forme “classique” d’AIH et la plus fréquente dans le monde. En comparaison, l’AIH de type II est rare et touche principalement les patients pédiatriques (filles/jeunes femmes). L’apparition d’une HCA peut se produire en combinaison avec d’autres maladies auto-immunes du foie (appelées “syndromes de chevauchement”). Le plus souvent, l’HCA chevauche la cirrhose biliaire primitive positive à l’AMA (environ 88%), mais des cholangites sclérosantes primitives et des hépatites virales peuvent également être présentes simultanément.
Diagnostic
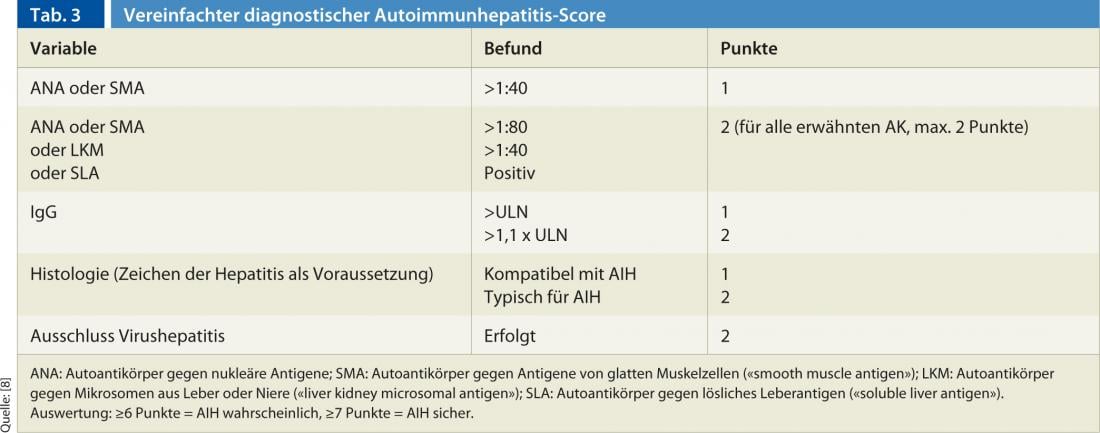
Le diagnostic repose sur une combinaison de résultats cliniques, histologiques et sérologiques, ainsi que sur l’exclusion d’autres hépatopathies chroniques (principalement les hépatites virales, les hépatites induites par les médicaments, les hépatopathies héréditaires et métaboliques) [7]. Aucun résultat individuel n’est pathognomonique de l’AIH. C’est pourquoi plusieurs scores ont été établis dans le passé, le score récemment publié et simplifié de Hennes et al. est facile à gérer dans la pratique clinique quotidienne [8]. Le score fait la distinction entre un diagnostic définitif (au moins 7 points) et un diagnostic probable (au moins 6 points) de l’AIH et se base sur les quatre critères suivants (tableau 3):
- Présence d’auto-anticorps
- Augmentation de l’immunoglobuline G (IgG)
- Histologie du foie
- Exclusion d’une hépatite virale
La sensibilité et la spécificité de ce score sont élevées : pour un diagnostic définitif, les valeurs sont respectivement de 81% et 99% ; pour un diagnostic probable, de 88% et 97% [8].
Laboratoire : Les analyses chimiques de laboratoire montrent généralement le tableau typique d’une atteinte hépatocellulaire : les transaminases sont nettement plus élevées que les paramètres de cholestase (AST : ratio ALP >3). Un tableau cholestatique avec hyperbilirubinémie directe prévalente et élévation de l’ALP est plus rare. En outre, si une HCA est suspectée, les gammaglobulines et les auto-anticorps contre les antigènes nucléaires (ANA), contre les antigènes des cellules musculaires lisses (“smooth muscle antigen” [SMA]) et contre les microsomes du foie ou du rein (“liver kidney microsomal antigen” [LKM]) doivent être déterminés.
Les nouveaux auto-anticorps supplémentaires sont par exemple les anticorps anti-cytosol hépatique 1 (LC-1), dont le titre est corrélé à l’activité inflammatoire de l’AIH, ou les anticorps anti-récepteur des asialoglycoprotéines (ASGPR). Ils peuvent être détectés dans jusqu’à 90% des AIH de type I et ont une signification pronostique.
Histologie : l’histologie est essentielle pour établir un diagnostic sûr, même si les résultats histologiques ne sont pas spécifiques de l’AIH [5]. Même si la présentation clinique est aiguë, des signes d’hépatopathie chronique sont généralement déjà présents à l’histologie. L’expression histologique n’est pas corrélée avec le degré d’augmentation des tests hépatiques ou des IgG. L’histologie est donc un paramètre plus fiable que les résultats de laboratoire pour évaluer la gravité de la maladie [9].
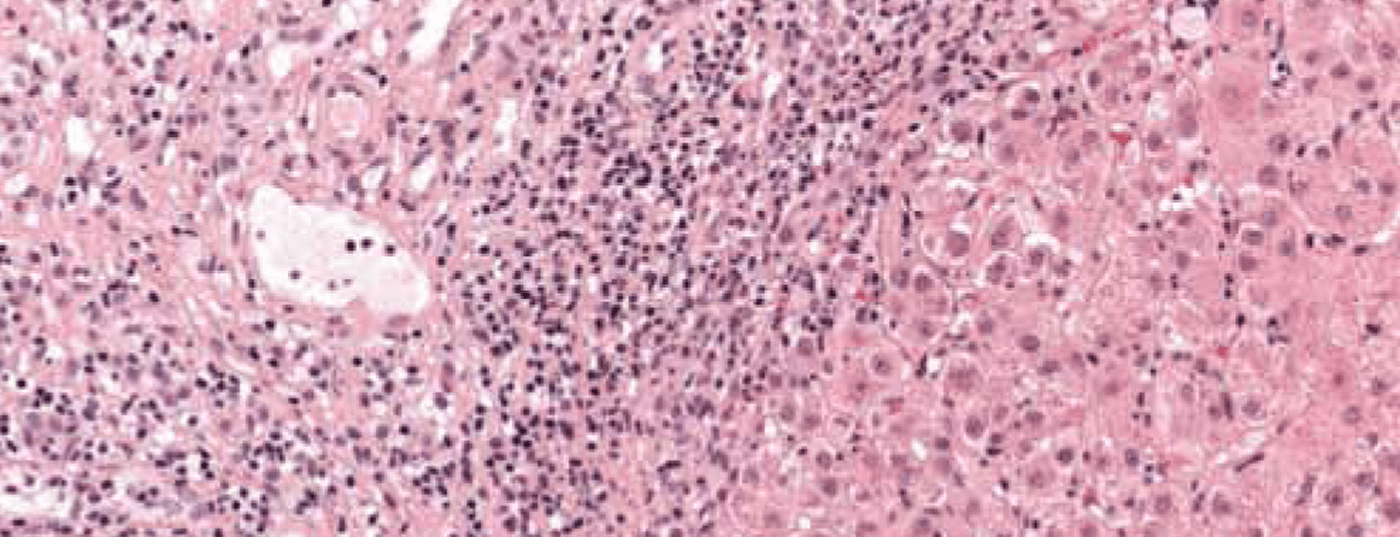
Le tableau caractéristique est celui d’une maladie chronique nécro-inflammatoire : infiltrats portaux et périportaux (“nécrose piecemeal”, “interface hepatitis”) et monocytaires (lymphoplasmocytaires) (figure 1). En cas de maladie grave et avancée, il existe une “bridging necrosis” étendue et une fibrose. Une “interface hépatite” n’indique pas nécessairement une évolution progressive de la maladie, par contre les “bridging necrosis” augmentent la probabilité de progression vers la cirrhose. La spécificité du résultat histologique global est de 81% et la valeur prédictive positive de 68%. Il est donc parfois difficile de distinguer une hépatite médicamenteuse ou virale d’une HCA.
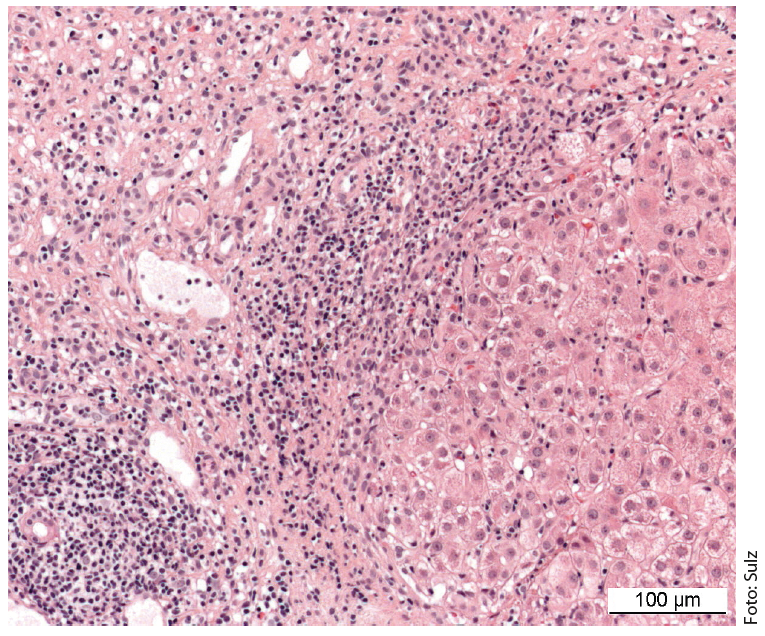
Fig. 1 : L’histologie du foie d’une jeune fille de 12 ans montre des coupes du champ portal partiellement fibrosées avec des infiltrats inflammatoires lymphoplasmocytaires denses et une hépatite d’interface active. Coloration : hématoxyline-éosine.
Thérapie
Indications : Selon l’American Association for the Study of Liver Diseases (AASLD), il existe des indications absolues et relatives pour le traitement de l’HCA (tableau 4) [3].

Classification de la thérapie : la thérapie peut être divisée en différentes phases :
- Induction de la rémission
- Maintien de la rémission
- Diminution ou arrêt du traitement
- Traitement d’une récidive
Objectif thérapeutique : L’objectif du traitement est la rémission durable. Classiquement, on définit la rémission comme la baisse de l’AST <2 fois la norme supérieure et la normalisation des gammaglobulines [8]. Cependant, des études récentes indiquent qu’une normalisation complète des tests hépatiques, de l’histologie et des gammaglobulines devrait être l’objectif du traitement, car cela a amélioré le pronostic des patients traités [10, 11].
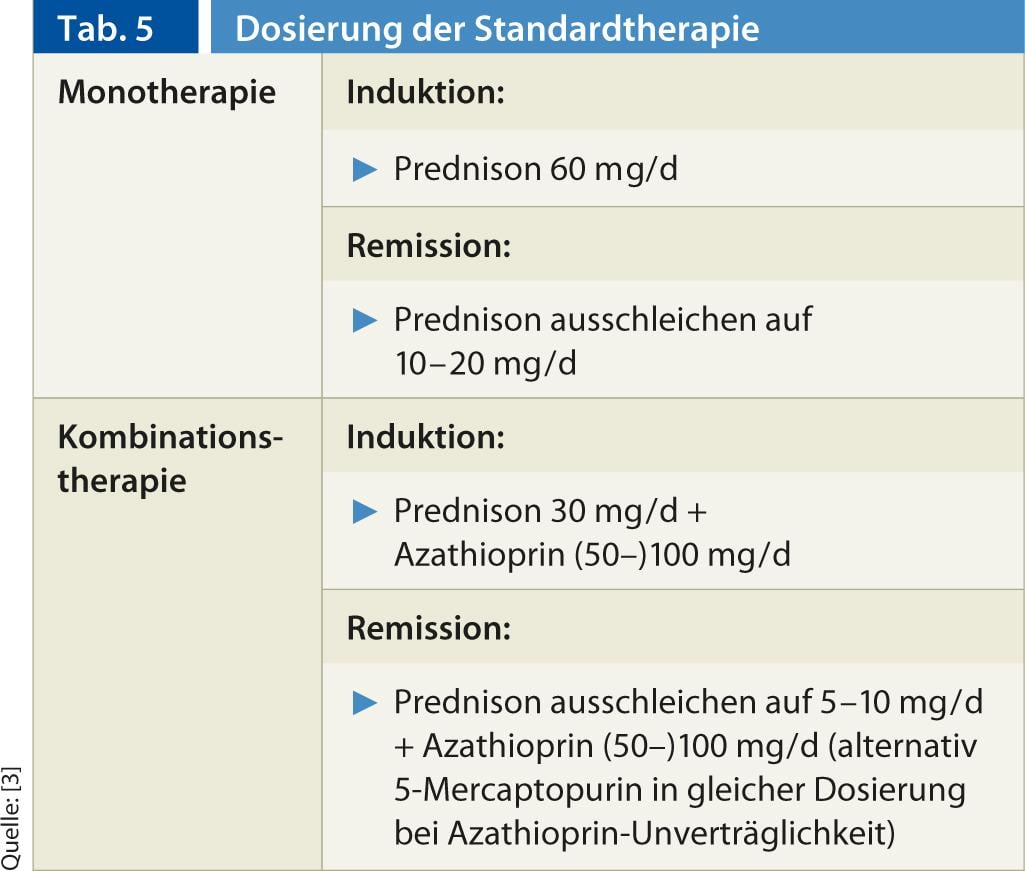
Traitement standard : pour le traitement immunosuppresseur de l’AIH, la collaboration du médecin de premier recours avec un spécialiste a fait ses preuves. Actuellement, le traitement standard consiste en une monothérapie ou une association de prednis(ol)-one avec/sans azathioprine, basée sur des études prospectives non randomisées menées entre les années 60 et le début des années 80 du siècle dernier. Les schémas posologiques sont mentionnés dans le tableau 5. Il est important de ne pas réduire trop rapidement la dose de stéroïdes. Il convient d’attendre la normalisation des transaminases. En cas de traitement combiné stéroïde/thiopurine, l’azathioprine est rapidement ajoutée au stéroïde. En cas d’intolérance à l’azathioprine, il est possible de passer à la 5-mercaptopurine (Puri-Nethol) à la même dose. L’efficacité de la thérapie combinée et de la monothérapie est comparable, mais la thérapie combinée est économe en stéroïdes, associée à des effets secondaires moins graves liés aux stéroïdes (de 66% à <20% pendant 18 mois de traitement) [12].
Traitement alternatif au budésonide : la plus grande étude thérapeutique contrôlée sur l’HCA réalisée à ce jour a comparé la valeur du budésonide (3 × 3 mg) à celle de la prednis(ol)-one en combinaison avec l’azathioprine [13]. Dans les deux bras de l’étude, on a observé un taux relativement faible de rémissions complètes, mais nettement moins d’effets secondaires liés aux stéroïdes dans le groupe budésonide (28% contre 53%). Le budésonide (2 × 3 mg) semble également bien adapté au maintien de la rémission.
Cependant, l’hypertension portale ou la cirrhose sont des contre-indications relatives au budésonide en raison de la réduction de son métabolisme [14].
Rémission et importance des récidives pour une durée de traitement suffisante : une rémission est obtenue dans 65 à 75% des cas après 24 mois de traitement. Environ 20% ne parviennent pas à une rémission complète. Une récidive (augmentation des transaminases et/ou des symptômes pendant le traitement ou pendant le sevrage ou après l’arrêt du traitement) survient dans environ 50% des cas dans les six mois ou dans 80% des cas après trois ans après l’arrêt du traitement, associée à une progression vers une cirrhose dans près de 40% et au développement d’une insuffisance hépatique dans 14%.
La durée du traitement avant l’arrêt de l’immunosuppression semble être le principal facteur permettant de distinguer les patients en rémission continue de ceux qui ont récidivé. Comparé au taux de rémission durable de 67% pour un traitement >4 ans, ce taux n’est que de 10% pour un traitement de 1-2 ans [15]. Il est donc recommandé de n’arrêter le traitement que sur la base d’une rémission clinique, histologique et biochimique complète après au moins deux ans, voire quatre ans. La rémission histologique peut être retardée jusqu’à huit mois par rapport à la rémission clinique et biochimique.
Options thérapeutiques plus récentes (“seconde ligne”) : Les options thérapeutiques alternatives doivent être discutées dans les cas suivants :
- Échec thérapeutique sous traitement standard
- Intolérance au traitement standard
- Éviter les effets secondaires des stéroïdes chez les patients à haut risque
- Études expérimentales
Les médicaments utilisés sont le mycophénolate, le mofétil (MMF), le tacrolimus ou la ciclosporine A, entre autres.
Transplantation hépatique : en cas d’échec du traitement, une transplantation hépatique doit être envisagée rapidement. Après une transplantation réussie, la survie est bonne (survie à 5 ans 91% [16]; survie à 10 ans 75% [cirrhose : 62%]). Le risque de récidive dans le greffon est de 20 à 36% [16] et est encore plus fréquent chez les enfants.
Surveillance du traitement et suivi : les transaminases et le taux d’IgG indiquent le succès du traitement et doivent être déterminés régulièrement (tous les trois à six mois). La détermination des auto-anticorps classiques au cours de l’évolution n’a pas de sens, car le niveau de titre n’est pas corrélé au niveau d’activité. Une biopsie du foie est recommandée avant l’arrêt du traitement, car une réaction inflammatoire résiduelle dans le foie peut déjà indiquer un risque accru de récidive.
En présence d’une cirrhose, il est recommandé de procéder à un dépistage du carcinome hépatocellulaire. En outre, les patients atteints d’HCA doivent être vaccinés contre l’hépatite A/B et recevoir les vaccinations standard habituelles [17].
Situations thérapeutiques particulières
Les patients à risque : Les risques d’effets secondaires du traitement sont les suivants : état métabolique diabétique, ostéoporose, hypertension artérielle mal contrôlée, instabilité émotionnelle ou antécédents positifs de psychose. Ces situations à risque ne constituent pas une contre-indication absolue, mais les patients doivent être suivis de près et les substances épargnant les stéroïdes, comme l’azathioprine, devraient être utilisées plus volontiers.
Hépatite fulminante : alors qu’une petite étude n’a pas montré de bénéfice pertinent des stéroïdes en cas d’HIA fulminante, la réponse aux stéroïdes en cas de présentation sévère était de 36 à 100 % dans d’autres études. En général, en cas de présentation fulminante, il est recommandé d’adresser immédiatement le patient à un centre de transplantation [18, 19].
Cirrhose : les patients avec et sans cirrhose présentent un bon résultat comparable (décès et transplantation hépatique comme critères d’évaluation) à dix ans (survie à 10 ans >90%), après quoi la courbe de survie des patients atteints de cirrhose (survie à 20 ans <40%) chute de manière significative par rapport aux patients non cirrhotiques (survie à 20 ans <80%).
Cirrhose décompensée : alors que le bénéfice du traitement n’est pas clair en cas de cirrhose histologiquement inactive, la réponse au traitement peut être très bonne en cas de cirrhose décompensée avec hépatite active. Une régression partielle de la fibrose est également possible [20].
Prévisions
Les patients atteints d’HCA et traités de manière adéquate ont une espérance de vie globalement normale et une qualité de vie bien préservée. Toutefois, environ ¹⁄3 des patients atteints d’HCA présentent déjà une cirrhose complète au moment du diagnostic initial, avec un pronostic moins bon en conséquence. A l’exception des évolutions fulminantes, la transplantation hépatique peut être évitée chez presque tous les patients non cirrhotiques et chez la plupart des patients atteints de cirrhose précoce.
Dr. Michael Christian Sulz
PD Dr. med. Tilman J. Gerlach
CONCLUSION POUR LA PRATIQUE
- L'(AIH) représente 11 à 23% de toutes les hépatopathies chroniques, mais sa fréquence est peut-être sous-estimée.
- La pathogenèse exacte de l’AIH reste incertaine. Le tableau clinique reflète une interaction complexe entre une prédisposition génétique, des facteurs déclencheurs, des auto-antigènes et des mécanismes d’immunorégulation, aboutissant finalement à un processus immunologique contre les hépatocytes.
- L’HCA a un aspect très hétérogène et fluctuant, avec différentes formes de manifestations allant de l’asymptomatique à l’hépatite aiguë, en passant par l’insuffisance hépatique aiguë et la cirrhose.
- Le diagnostic repose sur une combinaison de résultats cliniques, histologiques et sérologiques, ainsi que sur l’exclusion d’autres hépatopathies chroniques. Le score simplifié de Hennes et al. est facile à utiliser dans la pratique clinique quotidienne.
- Le traitement peut être divisé en différentes phases telles que l’induction, le maintien de la rémission, l’équilibrage ou l’arrêt et le traitement d’une récidive.
- L’objectif du traitement est la rémission durable. Le traitement standard est une monothérapie ou une association de prednis(ol)on avec/sans azathioprine.
Littérature :
- Boberg KM, et al : Scand J Gastroenterol 1998 ; 33 : 99-103.
- Milkiewicz P, et al : Transplantation 1999 ; 68 : 253-256.
- Manns MP, et al : Hepatology 2010 ; 51 : 2193-2213.
- Yeoman AD, et al : Hepatology 2009 ; 50 : 538-545.
- Czaja AJ, Freese DK : Hepatology 2002 ; 36 : 479-497.
- Sleisinger, Fordtran’s. : Maladies gastro-intestinales et hépatiques. 8e éd. Vol.2. Saunders, 2006.
- Krawitt EL. : N Engl J Med 2006 ; 354 : 54-66.
- Hennes EM, et al : Groupe international sur l’hépatite auto-immune. Hepatology 2008 ; 48 : 169-176.
- Czaja AJ, Wolf AM, Baggenstoss AH : Gastroenterology 1981 ; 80 : 687-692.
- Miyake Y, et al : J Hepatol 2005 ; 43 : 951-957.
- Montano-Loza AJ, Carpenter HA, Czaja AJ : Am J Gastroenterol 2007 ; 102 : 1005-1012.
- Summerskill WH, et al : Gut 1975 ; 16 : 876-883.
- Manns MP, et al : Gastroenterology. 2010 ; 139 : 1198-1206.
- Geier A, et al : World J Gastroenterol 2003 ; 9 : 2681-2685.
- Kanzler S, et al : J Hepatol 2001 ; 34 : 354-355.
- Campsen J, et al : Liver Transpl 2008 ; 14 : 1281-1286.
- Gleeson D, Heneghan MA : Gut 2011 ; 60 : 1611-1629.
- Kessler WR, et al : Clin Gastroenterol Hepatol 2004 ; 2 : 625-631.
- Czaja AJ : Liver Transpl 2007 ; 13 : 953-955.
- Dufour JF, DeLellis R, Kaplan MM : Ann Intern Med 1997 ; 127 : 981-985.
PRATIQUE DU MÉDECIN DE FAMILLE 2013 ; 5 : 10-14









