Le traitement chirurgical des patients atteints de maladies héréditaires du tissu conjonctif a pour objectif d’éviter la dissection aortique. En effet, celle-ci est responsable du taux de mortalité élevé de la population de patients. Quand l’indication est-elle donnée ? Et quelles sont les options thérapeutiques médicamenteuses ?
Les maladies héréditaires du tissu conjonctif avec des manifestations vasculaires sont peu présentes à l’esprit du clinicien généraliste. Pendant longtemps, le syndrome de Marfan a été le seul diagnostic différentiel dans ce sens chez les jeunes patients présentant un événement dans la région de l’aorte thoracique. Au cours de la dernière décennie, des centaines de gènes associés aux formes syndromiques et non syndromiques de la maladie aortique ont été identifiés [1].
Cependant, près de la moitié des patients ne sont diagnostiqués que dans le cadre d’une complication vasculaire. Alors que les anévrismes sont généralement détectés lors d’un examen de routine, la dissection aortique fait partie des urgences chirurgicales associées à une morbidité et une mortalité élevées.
Le syndrome de Marfan est une maladie héréditaire autosomique dominante dont l’incidence est d’environ 1/5000 naissances vivantes. Les manifestations oculaires, squelettiques et cardiaques typiques sont causées par des mutations du gène de la fibrilline 1, qui entraînent une suractivation de la voie de signalisation du TGFβ [2].
Il y a dix ans, Bart Loeys et Hal Dietz ont identifié une sous-population de patients qui se distinguaient par une luette fendue, un large écart entre les yeux et des vaisseaux sanguins tortueux. Entre-temps, différents gènes ont été identifiés, qui conduisent à un phénotype appartenant au groupe des syndromes de Loeys-Dietz. Il est important d’identifier ces patients, car les dissections sont fréquentes pour des diamètres aortiques qui n’étaient pas considérés jusqu’à présent comme une indication de remplacement prophylactique de l’aorte [3].
Le syndrome vasculaire rare d’Ehlers-Danlos est causé par des mutations du gène codant pour le collagène III. Ces patients se distinguent par leur taux élevé de dissections et de ruptures sans formation préalable d’anévrisme. Cela rend la prise en charge de ces patients très difficile. En l’absence de traitement, la survie moyenne est de 48 ans, les premiers événements survenant généralement au cours de la troisième ou de la quatrième décennie [4].
Options de traitement chirurgical
L’objectif du traitement chirurgical des patients atteints de maladies héréditaires du tissu conjonctif est d’éviter la dissection aortique due à une dilatation de l’aorte, car elle est responsable d’une mortalité élevée dans cette population de patients. Les directives de la Société européenne de cardiologie (ESC) de 2014 font clairement la distinction entre les patients atteints de Marfan ou de Loeys-Dietz et les patients sans maladie génétique sous-jacente pour poser l’indication d’un remplacement aortique électif [5]. La difficulté réside ici dans la délimitation. Chez les jeunes patients, on peut supposer la présence d’une composante génétique. Chez les patients sans maladie du tissu conjonctif, le remplacement aortique prophylactique est recommandé lorsque le diamètre de la racine aortique ou de l’aorte ascendante est de 55 mm. En revanche, les patients Marfan doivent être opérés à 50 mm, voire à partir de 45 mm en présence de facteurs de risque (comme des dissections dans la famille). Chez les patients atteints de la maladie de Loeys-Dietz, un remplacement électif est conseillé à 42-45 mm. En règle générale, le remplacement de la racine aortique est effectué en premier lieu. Dans la mesure du possible, on essaie de conserver la propre valve du patient. Si cela n’est pas possible en raison d’une lésion de la valve, un conduit supportant la valve est implanté.
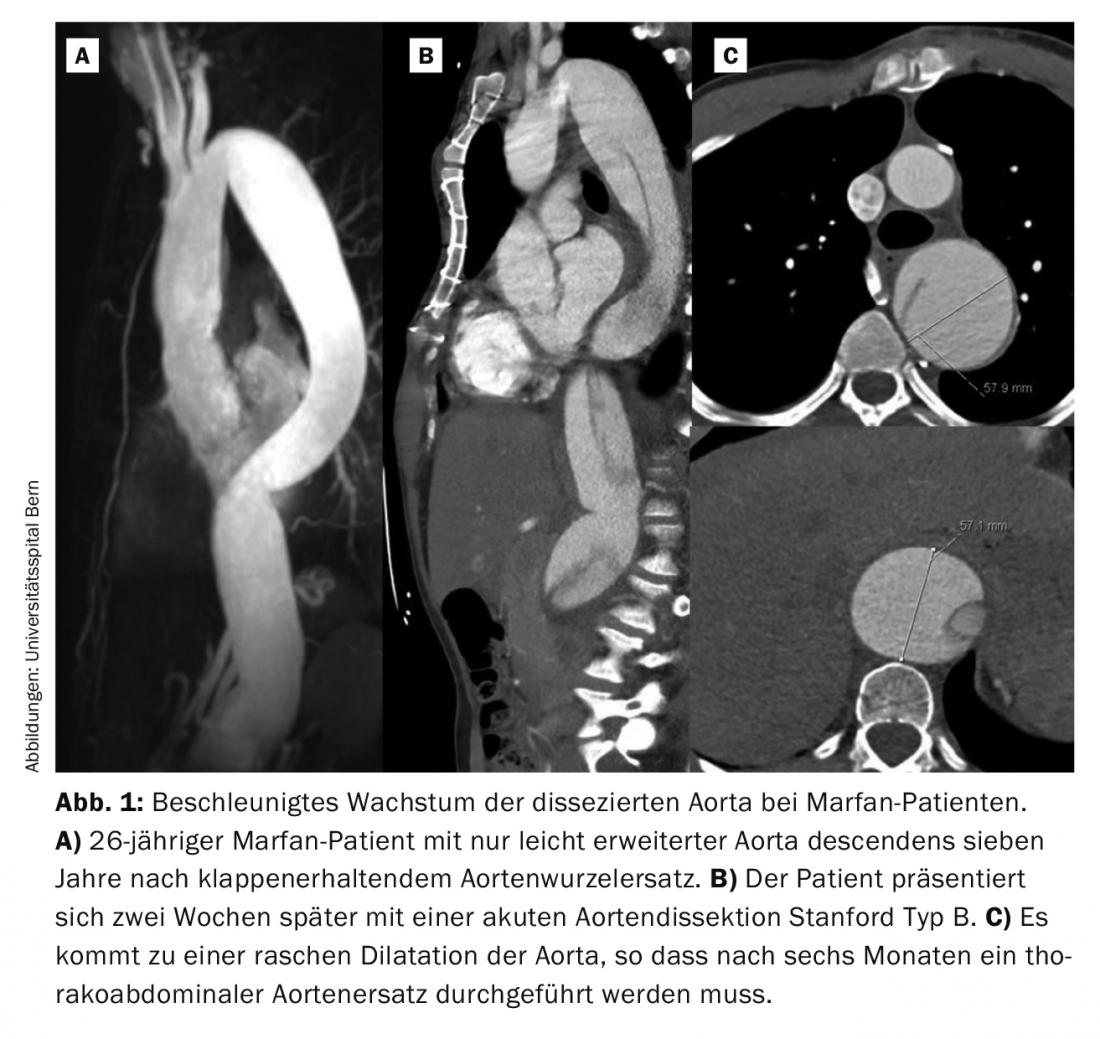
Cependant, la première manifestation est souvent la dissection aortique aiguë de Stanford de type A ou B. D’après notre expérience des vingt dernières années, un tiers des patients se sont présentés avec une dissection aiguë. La dissection aiguë de type A constitue une urgence chirurgicale et doit être traitée immédiatement. Même en cas d’opération réussie, la moitié de ces patients doivent être réopérés au cours de l’évolution, le plus souvent au niveau de l’aorte distale, c’est-à-dire non remplacée. Chez les patients ayant subi un remplacement électif de la racine aortique, seuls environ 10% des patients ont besoin d’une nouvelle intervention. Il s’agit dans la plupart des cas de patients ayant subi une dissection aortique de type B dans l’intervalle. Bien que la dissection aortique de type B soit souvent “non compliquée” au départ, c’est-à-dire sans malperfusion, l’aorte doit être remplacée ultérieurement par voie thoraco-abdominale chez la grande majorité des patients Marfan. La caractéristique typique des patients Marfan est une croissance rapide de l’aorte disséquée au cours des premières semaines et des premiers mois (Fig. 1). L’expérience montre que 50% des patients doivent être opérés dans l’année qui suit l’événement [6].
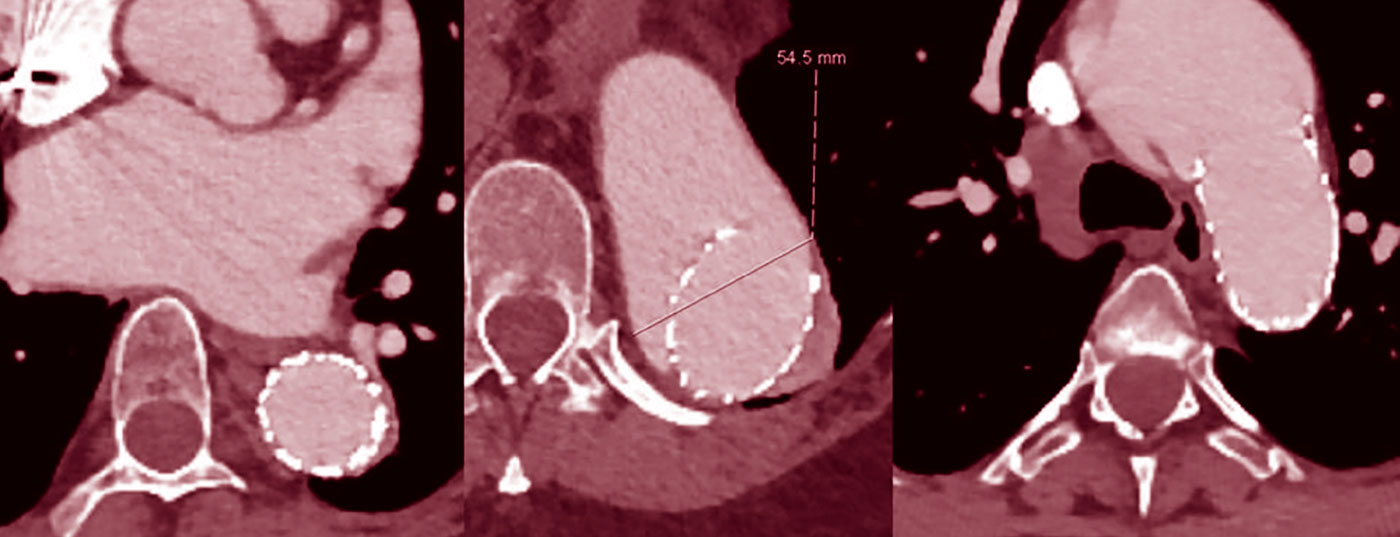
Le traitement des sections dilatées de l’aorte thoraco-abdominale par endoprothèses n’est généralement pas recommandé, car les sections vasculaires de la zone d’atterrissage se dilatent souvent secondairement (Fig. 2), même s’il y a un bon remodelage initial [7].

Options médicamenteuses
La dilatation et le risque de dissection de l’aorte qui en résulte sont la principale cause de morbidité et de mortalité chez les patients atteints de maladies héréditaires du tissu conjonctif. L’objectif du traitement médicamenteux est de réduire la progression de la dilatation et la survenue de dissections. La réduction de la pression artérielle à des valeurs systoliques maximales de 120 mmHg ainsi que la vitesse d’augmentation de la pression dans la région de l’aorte à chaque battement de cœur jouent un rôle important à cet égard. Le médicament de choix est traditionnellement le bêtabloquant [8]. Des études ont montré une vitesse de dilatation plus faible chez les patients sous traitement bêtabloquant, mais sans signification clinique en termes de survie [9].
Ces dernières années, le losartan, un bloqueur des récepteurs ATII, a été étudié plus en détail. Comme il intervient directement dans la voie de signalisation du TGFβ, on fonde de grands espoirs sur son action en matière de prévention de la dilatation et de la dissection aortiques [10]. Cependant, dans une grande étude randomisée chez des enfants atteints du syndrome de Marfan, aucune différence n’a été observée entre les bêtabloquants et le losartan [11]. L’étude a fait l’objet d’une controverse, car elle a finalement comparé une dose très élevée d’aténolol à une dose relativement faible de losartan. En raison de son profil d’effets secondaires plus faible, l’antagoniste ATII est donc chez nous le moyen de premier choix pour le traitement des patients sous traitement avec une fonction de pompe normale. Les antagonistes ATII sont particulièrement bien tolérés par les enfants et les adolescents. Des études à petite échelle ont montré que la pression artérielle n’était pas significativement réduite dans cette population de patients [12].
Suivi de l’évolution
Pour détecter à temps l’apparition ou la progression d’un anévrisme aortique et éviter une dissection aortique par un remplacement prophylactique précoce de l’aorte, il est essentiel d’effectuer un suivi étroit [13]. Dans ce cas, il est important d’évaluer systématiquement toutes les sections de l’aorte. En outre, il est important de procéder à des contrôles après une intervention sur l’aorte afin de détecter rapidement d’éventuelles complications telles que la formation d’anévrismes sur d’autres segments de l’aorte ou des dissections asymptomatiques.
Dans notre consultation, les patients sont suivis en postopératoire à trois et douze mois par angio-TDM, puis une à trois fois par an en fonction des résultats. Pour réduire la dose cumulée de radiations, cela devrait se faire principalement par IRM. L’examen IRM offre également l’avantage d’une imagerie fonctionnelle en ce qui concerne la fonction de pompage, les vices valvulaires et le diagnostic d’ischémie. Les opérations antérieures avec des implants posent rarement problème lors de l’évaluation des images. Pour l’évaluation des dissections, l’angiographie par scanner est actuellement encore supérieure à l’IRM. Des contrôles échocardiographiques sont effectués en cas de vitie ou de st.n. Remplacement valvulaire effectué une fois par an ou en alternance avec l’examen IRM.
Le désir d’enfant constitue une situation particulière dans le suivi à long terme des patients atteints de maladies héréditaires du tissu conjonctif. Il existe peu de preuves dans ce domaine et les recommandations des sociétés savantes sont parfois contradictoires [5,14,15]. On considère généralement qu’un diamètre aortique <40 mm présente un risque acceptable de grossesse sous contrôle échocardiographique étroit et bêtablocage. Pour les diamètres >45 mm, il est clairement conseillé de procéder à une opération prophylactique. Pour les diamètres compris entre 40 et 45 mm, la situation individuelle de la patiente doit être évaluée très soigneusement. Le facteur de risque ici est certainement des antécédents familiaux positifs en matière de dissection, en particulier pendant la grossesse.
Les directives actuelles recommandent le dépistage de tous les parents au premier degré d’un patient atteint d’un anévrisme de l’aorte thoracique. Cette étape permet de contribuer de manière significative à la réduction du taux de patients atteints de dissection.
Messages Take-Home
- Chez les patients atteints de maladies héréditaires du tissu conjonctif, l’indication d’un remplacement prophylactique de l’aorte est posée dès que le diamètre atteint 45-50 mm.
- Une évaluation régulière et systématique de toutes les sections de l’aorte permet d’éviter les dissections et les ruptures.
- Tous les parents au 1er degré de patients présentant un anévrisme de l’aorte thoracique doivent être examinés pour vérifier la présence d’un tel anévrisme.
Littérature :
- Brownstein AJ, et al : Gènes associés à l’anévrisme et à la dissection de l’aorte thoracique : mise à jour 2018 et implications cliniques. Aorte (Stamford) 2018 ; 6 : 13-20.
- Habashi JP, et al : Losartan, un antagoniste AT1, prévient l’anévrisme aortique dans un modèle murin du syndrome de Marfan. Science 2006 ; 312 : 117-121.
- Loeys BL, et al : Syndromes anévrismaux causés par des mutations dans le récepteur TGF-beta. N Engl J Med 2006 ; 355 : 788-798.
- Pepin M, et al : Caractéristiques cliniques et génétiques du syndrome d’Ehlers-Danlos de type IV, le type vasculaire. N Engl J Med 2000 ; 342 : 673-680.
- Erbel R, et al. : 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases : Document couvrant les maladies aortiques aiguës et chroniques de l’aorte thoracique et abdominale de l’adulte. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014 ; 35 : 2873-2926.
- Schoenhoff FS, et al : La dissection aortique aiguë détermine le devenir des segments aortiques initialement non traités dans le syndrome de Marfan. Circulation 2013 ; 127 : 1569-1575.
- Grabenwöger M, et al : Thoracic Endovascular Aortic Repair (TEVAR) for the treatment of aortic diseases : a position statement from the European Association for Cardio- Thoracic Surgery (EACTS) and the European Society of Cardiology (ESC), in collaboration with the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2012 ; 33 : 1558-1563.
- Shores J, et al : Progression de la dilatation aortique et bénéfice du blocage bêta-adrénergique à long terme dans le syndrome de Marfan. N Engl J Med 1994 ; 330 : 1335-1341.
- Gersony DR, et al : The effect of beta-blocker therapy on clinical outcome in patients with Marfan’s syndrome : a meta-analysis. Int J Cardiol 2007 ; 114 : 303-308.
- Habashi JP, et al : La signalisation du récepteur de l’angiotensine II de type 2 atténue l’anévrisme aortique chez la souris par l’intermédiaire de l’antagonisme ERK. Science 2011 ; 332 : 361-365.
- Lacro RV, et al : Aténolol versus losartan chez les enfants et les jeunes adultes atteints du syndrome de Marfan. N Engl J Med 2014 ; 371 : 2061-2071.
- Brooke BS, et al : Blocage de l’angiotensine II et dilatation de l’aorte dans le syndrome de Marfan. N Engl J Med 2008 ; 358 : 2787-2795.
- Jondeau G, et al : Taux d’événements aortiques dans la population marfan une étude de cohorte. Circulation 2012 ; 125 : 226-232.
- Baumgartner H, et al : ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J 2010 ; 31 : 2915-2957.
- Hiratzka LF, et al.: 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with Thoracic Aortic Disease : a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010 ; 121 : e266-369.
CARDIOVASC 2018 ; 17(5) : 27-29









