La thérapie génique, longtemps porteuse d’espoir pour la recherche médicale, trouve enfin le chemin de la clinique pour guérir les maladies monogéniques. La médecine dispose ainsi d’une nouvelle thérapie qui peut traiter les maladies héréditaires non seulement de manière symptomatique, mais aussi de manière causale. Une autre approche de thérapie génique consiste à traiter les altérations génétiques acquises dans les tumeurs malignes à l’aide de cellules T CAR (chimeric antigen receptor).
La thérapie génique, longtemps porteuse d’espoir pour la recherche médicale, trouve enfin le chemin de la clinique pour guérir les maladies monogéniques. La médecine dispose ainsi d’une nouvelle thérapie qui peut traiter les maladies héréditaires non seulement de manière symptomatique, mais aussi de manière causale. Une autre approche de thérapie génique consiste à traiter les altérations génétiques acquises dans les tumeurs malignes à l’aide de cellules T CAR (chimeric antigen receptor). Les paragraphes suivants donnent un aperçu des bases de la biologie moléculaire et de l’application clinique des techniques courantes de thérapie génique et cellulaire.
Les gènes comme plans de toutes les protéines – les mutations comme cause des maladies monogéniques
Chaque cellule humaine contient un plan de construction génétique à l’intérieur du noyau cellulaire sous la forme d’acide désoxyribonucléique (ADN). L’ADN est composé de quatre éléments de base, les bases adénine (A), thymine (T), guanine (G) et cytosine (C), qui, alignées en longues chaînes linéaires, forment la structure de base des chromosomes d’une cellule humaine. L’ADN de chaque cellule contient des unités codantes individuelles, les gènes. Chaque gène est composé d’une région codante et d’une unité régulatrice, le promoteur, dont l’activité peut éventuellement être renforcée par un élément enhancer. Chaque gène code pour la construction d’une protéine en suivant l’ordre des quatre éléments de base de l’ADN. Les protéines sont composées de 20 éléments de base, les acides aminés, qui se replient les uns sur les autres en une ou plusieurs chaînes linéaires pour former des protéines. Seules les protéines correctement repliées peuvent exercer leur fonction biologique dans la cellule.
Maladies monogéniques
Si une réparation défectueuse se produit après l’endommagement de l’ADN, l’ordre des éléments de base de l’ADN peut être modifié. Alternativement, des éléments de base de l’ADN peuvent être perdus. Ces changements sont appelés “mutations”. Si une mutation se produit dans un segment d’ADN codant pour une protéine, la structure de la protéine codée peut être modifiée : La protéine défectueuse synthétisée selon le blueprint de l’ADN ne peut pas remplir sa fonction naturelle dans la cellule, ou seulement de manière incomplète. Cette modification génétique, dans le cas le plus simple, par exemple le remplacement d’un A par un C au sein de l’ADN, peut être la cause causale d’une maladie d’origine génétique si elle entraîne une diminution, voire une disparition, de la fonction de la cellule dans un tissu, comme par exemple
- lorsque le métabolisme cellulaire ne fonctionne pas suffisamment et que les métabolites s’accumulent (par exemple, défaut de lipoprotéine lipase)
- lorsque la synthèse de certains éléments de base par le métabolisme est déficiente (par ex. défaut de glucose-6-phosphate déshydrogénase)
- lorsque certains composants du système immunitaire ne fonctionnent pas (par exemple, granulomatose septique ou déficit immunitaire combiné sévère)
Une étude récente a permis d’établir un lien de causalité entre 4166 maladies monogéniques rares et 3163 gènes [1]. La guérison de la maladie d’origine génétique n’est possible que si l’on parvient à compenser ou à corriger la cause causale.
Nouvelles approches de guérison par thérapie génique
Lorsqu’une mutation génétique clairement définie a été identifiée comme étant la cause d’une maladie, cette maladie devient accessible à une thérapie génique potentielle. Différentes procédures sont disponibles :
- Dans les maladies où la mutation génétique entraîne une perte d’expression de la protéine ou l’expression d’une protéine défectueuse (mutations “loss of function” ou “dysfunction”), une addition de gènes peut être effectuée.
- Dans les maladies où la mutation du gène entraîne une hyperfonction de la protéine (mutations “gain de fonction”), une réparation du gène peut être envisagée.
L’addition de gènes consiste à ajouter au génome de la cellule un gène qui code correctement la protéine manquante ou qui fonctionne mal, compensant ainsi la fonction du gène muté. Il en résulte une restauration de la fonction initiale de la protéine. L’addition de gènes a été testée dans le cadre d’essais cliniques pour le traitement de nombreuses maladies, avec des succès cliniques pour plus de 20 maladies héréditaires congénitales. Tous les produits de thérapie génique actuellement autorisés sont basés sur l’addition de gènes.
La réparation génique à l’aide de nucléases telles que CRISPR-Cas9 (fusion de la nucléase Cas9 avec CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats, qui se lie à l’ADN) permet de rétablir la séquence d’ADN correcte dans la cellule en ciblant la mutation et en la corrigeant in situ (principe : exciser et remplacer). Ainsi, la fonction prévue de la protéine est restaurée et la fonction biologique est normalisée par la suite. L’utilisation de la réparation génétique (“ciseaux génétiques” /CRISPR-Cas) est possible indépendamment de la présence de mutations “gain de fonction” ou “perte de fonction”. Une telle modification de l’information génétique spécifique à la séquence est possible en laboratoire, mais n’a été utilisée jusqu’à présent qu’en oncologie dans le cadre de quelques études cliniques.
Addition de gènes
Les premiers succès cliniques de la thérapie génique ont été obtenus dans le domaine de l’immunologie. Les causes des déficits immunitaires congénitaux se trouvent dans les cellules souches hématopoïétiques. Les patients concernés peuvent donc souvent être guéris par une transplantation de cellules souches hématopoïétiques provenant d’un donneur étranger ou familial (transplantation allogénique). Les membres de la famille sont rarement utilisés comme donneurs haploïdes HLA en raison d’un taux d’effets secondaires plus élevé. Les membres de la famille qui ont une identité HLA plus élevée, mais qui sont également porteurs du défaut génétique, sont plus susceptibles de ne pas être éligibles. En cas d’absence de donneur HLA-ident (dans environ 1/3 des cas pour les Caucasiens, plus souvent pour les autres ethnies), une thérapie génique autologue peut conduire à la guérison de la maladie.
Pour la thérapie génique ex vivo, les cellules autologues sont aphérisées, purifiées, enrichies et cultivées ex vivo pendant une courte période après avoir été mobilisées avec du G-CSF. Pendant cette période, les cellules souches sont traitées avec le vecteur de thérapie génique et sont ensuite réinjectées au patient, généralement après une chimiothérapie, pour l’ablation de la moelle osseuse. Selon le déficit immunitaire, une correction partielle peut, de manière surprenante, être suffisante pour une amélioration clinique significative.
La première publication sur une thérapie génique réussie a été publiée en 2000 sur le traitement des nourrissons atteints de la forme la plus grave d’une immunodéficience congénitale, l’immunodéficience combinée sévère, qui, en l’absence de transplantation, entraîne généralement la mort au cours de la première année de vie en raison d’infections très graves [2].
Entre 2000 et 2006, tous les succès cliniques dans le domaine de la thérapie génique ont été obtenus grâce à l’utilisation de vecteurs dits rétroviraux. Ces derniers introduisent la séquence de correction sous forme d’ARN dans les cellules souches, où elle est convertie en ADN par transcription inverse, puis intégrée dans l’ADN du patient (addition de gènes). Une protéine fonctionnelle est ensuite produite, ce qui permet de corriger cliniquement le défaut.
Pour une addition de gènes in vivo, les cellules du corps ne sont pas prélevées, traitées et restituées, mais des particules virales avec une séquence de correction sont injectées directement dans le corps. La première addition de gènes in vivo a été publiée en 2007 : pour traiter la maladie de Parkinson, des particules virales adéno-associées (AAV) modifiées ont été injectées unilatéralement, par voie sous-thalamique, à des patients et ont codé pour l’acide glutamique décarboxylase [3]. Dans la grande majorité des essais cliniques de thérapie génique in vivo, des particules d’AAV modifiées sont utilisées pour introduire des informations génétiques dans l’organisme. Les exceptions sont l’utilisation de particules virales modifiées basées sur le virus de l’herpès simplex 1 (HSV-1) [4] ou de particules adénovirales non réplicatives [5] pour le traitement du glioblastome, ainsi que les essais de particules virales dérivées du VIH-1 pour le traitement in vivo de la maladie de Parkinson [6].
Effets secondaires dans les premiers essais de thérapie génique
La première génération de thérapies géniques ex vivoutilisant des vecteurs γ-rétroviraux a montré que le lieu d’intégration du vecteur de thérapie génique dans le génome de la cellule cible joue un rôle clé en ce qui concerne les effets secondaires potentiels. Ces vecteurs de première génération s’intègrent de manière préférentielle à proximité des sites d’initiation de la transcription. Dans les vecteurs de thérapie génique γ-rétrovirale de première génération, l’expression de la protéine était entraînée par un promoteur dans le vecteur de thérapie génique et renforcée par un amplificateur. L’amplificateur du vecteur de thérapie génique de première génération pouvait interagir avec le site d’initiation de la transcription du gène dans lequel le vecteur de thérapie génique était intégré. Si l’intégration dans un oncogène a eu lieu, cet oncogène a pu être activé en plus de l’expression thérapeutique de la protéine qui a conduit à la guérison de la maladie de base. Les cellules souches dans lesquelles cette constellation était présente avaient ainsi acquis un avantage de croissance et pouvaient se multiplier de manière clonale. Dans certains essais cliniques portant sur des déficits immunitaires divers, cela a eu pour conséquence que certains patients ont développé des hémopathies malignes comme effet secondaire de la thérapie génique [7–10]. Ces effets secondaires ont conduit, à partir de 2009, à l’abandon des vecteurs γ-rétroviraux au profit de vecteurs lentiviraux dérivés du virus VIH-1, dont le profil d’intégration promet une plus grande sécurité [11]. En outre, dans les vecteurs lentiviraux auto-activants (SIN), le promoteur issu du virus VIH-1 et son élément amplificateur ont été supprimés. L’absence d’éléments enhancers dans les vecteurs de thérapie génique de deuxième et troisième génération empêche la transactivation des oncogènes médiée par les enhancers après le traitement ex vivodes cellules souches. À ce jour, on estime que les cellules souches d’une centaine de patients ont été traitées avec des vecteurs lentiviraux de la NAS dans le cadre d’essais cliniques. Jusqu’à présent, aucune de ces études cliniques n’a révélé de malignité hématologique, ce qui indique une amélioration significative de la sécurité par rapport à la première génération de vecteurs.
Les vecteurs de thérapie génique basés sur l’AAV utilisés pour l’addition de gènes in vivone s’intègrent que de manière limitée dans le génome des cellules cibles. Par conséquent, une mutagenèse par insertion est peu probable. Le plus grand risque potentiel des vecteurs de thérapie génique basés sur les AAV réside dans une éventuelle immunité préexistante contre les structures de surface du vecteur AAV. Dans une étude clinique sur le traitement de l’hémophilie B, un vecteur AAV2 codant pour le facteur IX de coagulation a été utilisé par voie intramusculaire. Une réponse immunitaire médiée par les cellules T a conduit à l’élimination de toutes les cellules génétiquement modifiées et a mis fin à l’effet thérapeutique [12]. En revanche, l’administration intraveineuse a entraîné l’absorption des vecteurs par les hépatocytes et des effets thérapeutiques à long terme chez les patients chez lesquels aucun anticorps anti-AAV n’avait été détecté avant l’inclusion dans l’étude [13].
Succès de la thérapie génique
La première génération de vecteurs de thérapie génique γ-rétrovirale a apporté la preuve que la thérapie génique peut conduire à des succès cliniques pour le X-SCID (déficit immunitaire combiné sévère lié à l’X) [2], le SCID ADA (adénosine désaminase) [14], le X-CGD (granulomatose septique liée à l’X) [9], l’épidermolyse bulleuse [15] et le syndrome de Wiskott Aldrich (WAS) [16].
En raison des effets secondaires susmentionnés lors du traitement de l’X-SCID, de l’X-CGD et du WAS avec des vecteurs de thérapie génique de première génération (vecteurs γ-rétroviraux à promoteur/enhancer complet), des vecteurs lentiviraux SIN ont été développés et ont donné lieu à une réussite clinique. Jusqu’à présent, les vecteurs lentiviraux du SIN ont permis d’obtenir des succès cliniques dans la thérapie génique ex vivo pour le traitement de
- adrénoleucodystrophie liée au chromosome X (ALD) [17]
- γ-Thalassémie [18,19]
- QUOI [20]
- X-SCID [21]
- ADA-SCID [22]
- Maladie de la drépanocytose [23]
- COL7A1 Epidermolyse bulleuse [24]
- Leucodystrophie métachromatique [25]
La thérapie génique in vivomédiée par l’AAV a également été utilisée pour traiter au moins 14 indications :
Neurologie
- Maladie de Parkinson [3,26,27]
Ophtalmologie
- Cong. de foie Amaurose [28,29]
- Choroïdérémie [30,31]
- Dégénérescence maculaire liée à l’âge [32]
- Neuropathie optique héréditaire de Leber [33,34]
Hématologie
- Hémophilie B [13]
Dystrophie musculaire
- Dystrophie musculaire de Becker [35]
- Atrophie musculaire spinale de type I (SMA1) [36]
Maladies métaboliques
- Maladie de Pompe [37]
- Déficit en α-1-antitrypsine (AAT) [38,39]
- Mucopolysaccharidose de type IIIB [40]
- Acide aminé aromatique Décarboxylase déficiente [41,43]
- Déficit en lipoprotéine lipase [42]
Dans le traitement de la leucémie, les cellules CAR-T sont utilisées dans de nombreuses études et dans des thérapies désormais approuvées. Pour ce faire, des récepteurs antigéniques chimériques sont introduits dans des cellules T autologues de patients atteints. Cela permet à ces cellules T génétiquement modifiées (cellules CAR-T) de reconnaître et d’éliminer les cellules tumorales [44–46].
Réparation du genre
Jusqu’à présent, il n’est pas possible de faire intégrer des vecteurs de thérapie génique lenti- ou γ-rétroviraux à des endroits prédéterminés du génome. Une insertion et/ou une correction spécifique à la séquence n’a été possible qu’après avoir réussi à couper l’ADN de manière spécifique à la séquence. Cela se fait au moyen de nucléases telles que CRISPR-Cas. Dans ce système de thérapie génique, une fusion a été réalisée entre le CRISPR ( Clustered Regularly Interspaced Short Palindromic Repeats ), qui se lie à l’ADN, et le Cas9 (CRISPR/Cas9), qui coupe l’ADN : [47]. La cassure de l’ADN qui en résulte peut alors être réparée par la cellule de deux manières différentes : Soit les extrémités sont reconnectées par un processus défectueux appelé jonction d’extrémité non homologue. Le deuxième mécanisme de réparation est la recombinaison homologue. Cette voie est empruntée lorsqu’il existe dans la cellule un ADN de réparation dont les extrémités correspondent à la séquence d’ADN à l’endroit de la cassure de l’ADN. Cet ADN de réparation est administré aux cellules en même temps que la nucléase et sert de modèle de correction génétique (template). Lors de la recombinaison homologue, le site de cassure de l’ADN est alors fermé par les enzymes de réparation cellulaires endogènes, conformément à la séquence d’ADN de l’ADN de réparation. Il est ainsi possible d’introduire dans le génome un gène de correction spécifique à la séquence ou de corriger génétiquement des mutations ponctuelles. L’addition de gènes spécifiques à une séquence n’a pas encore trouvé d’application clinique, car l’efficacité de la méthode était jusqu’à présent limitante. Cependant, les récents développements technologiques rendent les essais cliniques possibles.
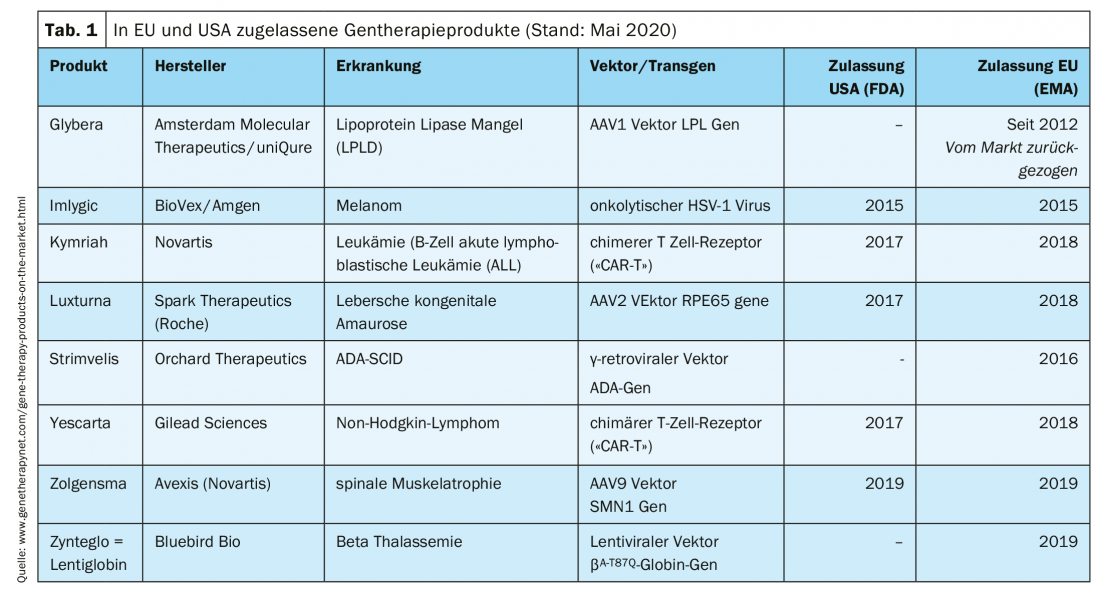
Produits de thérapie génique cliniquement approuvés
Le traitement des patients avec des produits de thérapie génique est possible dans des centres spécialisés. Plusieurs produits ont déjà reçu une autorisation de mise sur le marché (tableau 1).
Messages Take-Home
- La thérapie génique en tant que traitement causal des maladies héréditaires monogéniques trouve de plus en plus sa place dans les cliniques.
- La thérapie génique par vecteurs viraux, basée sur l’addition de gènes, est utilisée avec succès dans des études cliniques depuis 2000 pour certaines maladies hématologiques.
- et les déficits immunitaires congénitaux.
- La réparation ciblée des gènes à l’aide de nucléases a été utilisée jusqu’à présent dans quelques études pour des maladies oncologiques dans lesquelles la fonction d’un gène doit être supprimée.
- Certaines tumeurs malignes peuvent être traitées à l’aide de cellules T génétiquement modifiées (CAR-T cells).
- Les médicaments de thérapie génique sont actuellement déjà disponibles en tant que produits autorisés pour 7 maladies.
Littérature :
- Ehrhart F, et al. : Historique des maladies rares et de leurs causes génétiques – une approche basée sur les données. bioRxiv 2020 ; preprin doi : https://doi.org/10.1101/595819
- Cavazzana-Calvo, M et al : Thérapie génétique de l’immunodéficience humaine sévère combinée (SCID)-X1. Science 2000 ; 288 : 669-672.
- Kaplitt MG, et al : Sécurité et tolérance de la thérapie génique avec un gène Borne GAD du virus adéno-associé (AAV) pour la maladie de Parkinson : un essai de phase I à étiquette ouverte. Lancet 2007 ; 369(9579) : 2097-2105.
- Todo T : Immunothérapie active : thérapie virale oncolytique utilisant le HSV-1 Adv Exp Med Biol. 2012 ; 746 : 178-186.
- Brenner AJ, et al. : Sécurité et efficacité du VB-111, un traitement génétique anticancéreux, chez les patients atteints de glioblastome récurrent : résultats d’une étude de phase I/II. Neuro Oncol 2020 ; 22(5) : 694-704.
- Palfi S, et al : Suivi à long terme d’une étude de phase I/II de ProSavin, un traitement génétique vecteur lentiviral pour la maladie de Parkinson. Hum Gene Ther Clin Dev 2018 ; 29(3) : 148-155.
- Hacein-Bey-Abina S, et al : Serious Adverse Event After Successful Gene Therapy for X-linked Severe Combined Immunodeficiency. N Engl J Med 2003 ; 348(3) : 255-256.
- Aiuti A, et al : The Committee for Advanced Therapies’ of the European Medicines Agency Reflection Paper on Management of Clinical Risks Deriving from Insertional Mutagenesis. Human Gene Ther Clin Dev 2013 ; 24 : 47-54.
- Ott MG, et al : Correction de la maladie granulomateuse chronique liée à l’X par thérapie génique, augmentée par l’activation insertionnelle de MDS1-EVI1, PRDM16 ou SETBP1. Nat Med 2006 ; 12(4) : 401-409.
- Siler U, et al : Successful Combination of Sequential Gene Therapy and Rescue Allo-HSCT in Two Children With X-CGD – Importance of Timing. Curr Gene Ther 2015 ; 15(4) : 416-427.
- Serrao E, et al : Sites d’intégration de l’ADN rétroviral : de la recherche fondamentale aux applications cliniques. AN Crit Rev Biochem Mol Biol 2015 ; 28 : 1-17.
- Manno CS, et al : Transduction réussie du foie dans l’hémophilie par le facteur IX de l’AAV et limites imposées par la réponse immunitaire de l’hôte. Nat Med 2006 ; 12 : 342-347.
- Nathwani AC, et al : Long-term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med 2014 ; 371(21) : 1994-2004.
- Aiuti A, et al : Correction de l’ADA-SCID par la thérapie génique des cellules souches combinée à un conditionnement non myéloablatif. Science 2002 ; 296(5577) : 2410-2413.
- Mavilio F, et al : Correction de l’épidermolyse bulleuse jonctionnelle par transplantation de cellules souches épidermiques génétiquement modifiées. Nat Med 2006 ; 12(12) : 1397-1402.
- Boztug K, et al : Stem-cell Gene Therapy for the Wiskott-Aldrich Syndrome. N Engl J Med 2010 ; 363(20) : 1918-1927.
- Cartier N, et al : Thérapie génique des cellules souches hématopoïétiques avec un vecteur lentiviral dans l’adrénoleucodystrophie liée à l’X. Science 2009 ; 326(5954) : 818-823.
- Cavazzana-Calvo M, et al : Indépendance transfusionnelle et activation de HMGA2 après traitement génétique de la β-thalassémie humaine. Nature 2010 ; 467(7313) : 318-322.
- Thompson AA, et al : Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med 2018 ; 378 : 147993.
- Aiuti A, et al : Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013 ; 341(6148) : 1233151.
- De Ravin SS, et al : Lentiviral Hematopoietic Stem Cell Gene Therapy for X-linked Severe Combined Immunodeficiency. Sci Transl Med 2016 ; 8(335) : 335ra57.
- Mullard A : L’EMA approuve la thérapie génique secondaire. Nat Rev Drug Discov 2016 ; 15 : 299.
- Ribeil JA, et al : Thérapie génétique chez un patient atteint de la maladie drépanocytaire. N Engl J Med 2017 ; 376(9) : 848-855.
- Lwin SM, et al : Safety and Early Efficacy Outcomes for Lentiviral Fibroblast Gene Therapy in Recessive Dystrophic Epidermolysis Bullosa. JCI Insight 2019 ; 4(11) : e126243.
- Biffi A, et al : Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013 ; 341(6148) : 1233158.
- Niethammer M, et al : Gene Therapy Reduces Parkinson’s Disease Symptoms by Reorganizing Functional Brain Connectivity. Sci Transl Med 2018 ; 10(469) : eaau0713.
- Heiss JD, et al : Essai de thérapie génique putaminale guidée par résonance magnétique pour la maladie de Parkinson avancée. Mov Disord 2019 ; 34(7) : 1073-1078.
- Bainbridge JWB, et al : Effet de la thérapie génétique sur la fonction visuelle dans l’amaurose congénitale de Leber. N Engl J Med 2008 ; 358(21) : 2231-2239.
- Bainbridge JWB, et al : Effet à long terme de la thérapie génétique sur l’amaurose congénitale de Leber. N Engl J Med 2015 ; 372(20) : 1887-1897.
- MacLaren RE, et al : Retinal Gene Therapy in Patients With Choroideremia : Initial Findings From a Phase 1/2 Clinical Trial. Lancet 2014 ; 383 : 1129-1137.
- Xue K, et al : Beneficial Effects on Vision in Patients Undergoing Retinal Gene Therapy for Choroideremia. Nat Med 2018 ; 24(10) : 1507-1512.
- Rakoczy EP, et al : Gene Therapy With Recombinant Adeno-Associated Vectors for Neovascular Age-Related Macular Degeneration : 1 Year Follow-Up of a Phase 1 Randomised Clinical Trial. Lancet 2015 ; 386(10011) : 2395-2403.
- Feuer WJ, et al : Thérapie génétique pour la neuropathie optique héréditaire de Leber : Résultats initiaux. Ophthalmology 2015 ; 123(3) : 558-570.
- Bouquet C, et al : Réponse immunitaire et inflammation intraoculaire chez les patients atteints de neuropathie optique héréditaire de Leber traités par injection intravitréenne de virus adéno-associé recombinant 2 portant le gène ND4 : une analyse secondaire d’un essai clinique de phase 1/2. JAMA Ophthalmol 2019 ; 137(4) : 399-406.
- Mendell JR, et al : A Phase 1/2a Follistatin Gene Therapy Trial for Becker Muscular Dystrophy. Mol Ther 2015 ; 23(1) : 192-201.
- Mendell JR, et al : Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 2017 ; 377(18) : 1713-1722.
- Corti M, et al : La déplétion des cellules B est protectrice contre la réponse immunitaire anti-AAV Capsid : une étude de cas sur un sujet humain. Mol Ther Methods Clin Dev 2014 ; 1 : 14033.
- Calcedo R, et al : Réponses des cellules T de classe I à un peptide polymorphe dans un essai clinique de thérapie génique pour le déficit en α-1-antitrypsine. Proc Natl Acad Sci U S A 2017 ; 114(7) : 1655-1659.
- Mueller C, et al : Expression de l’année et réparation des défauts des neutrophiles après thérapie génique dans le déficit en alpha-1 antitrypsine. Mol Ther 2017 ; 25(6) : 1387-1394.
- Tardieu M, et al : Intracérébral Gene Therapy in Children With Mucopolysaccharidosis Type IIIB Syndrome : An Uncontrolled Phase 1/2 Clinical Trial. Lancet Neurol 2017 ; 16(9) : 712-720.
- Chien YH, et al : Efficacité et sécurité de la thérapie génique AAV2 chez les enfants souffrant d’une déficience en L-amino acide décarboxylase aromatique : un essai de phase 1/2, à label ouvert. Lancet Child Adolesc Health 2017 ; 1(4) : 265-273.
- Kassner U, et al : Gene Therapy in Lipoprotein Lipase Deficiency : Case Report on the First Patient Treated With Alipogene Tiparvovec Under Daily Practice Conditions. Hum Gene Ther 2018 ; 29(4) : 520-527.
- Kojima K, et al : La thérapie génique améliore les fonctions motrices et mentales des personnes souffrant d’un déficit en L-amino acide décarboxylase aromatique. Brain 2019 ; 142(2) : 322-333.
- Porter DL, et al : Cellules T modifiées par des récepteurs antigéniques chimériques dans la leucémie lymphoïde chronique. N Engl J Med 2011 ; 365(8) : 725-733.
- Savoldo B, et al : La costimulation du CD28 améliore l’expansion et la persistance des cellules T modifiées par des récepteurs antigéniques chimériques chez les patients atteints de lymphome. J Clin Invest 2011 ; 121(5) : 1822-1826.
- Grupp SA, et al : Cellules T modifiées par des récepteurs antigéniques chimériques pour la leucémie lymphoïde aiguë. N Engl J Med 2013 ; 368(16) : 1509-1518.
- Yang G, Huang X : Méthodes et applications du système CRISPR/Cas pour l’édition du génome dans les cellules souches. Cell Regen (Lond) 2019 ; 8(2) : 33-41.
PRATIQUE DU MÉDECIN DE FAMILLE 2020 ; 15(9) : 6-10









