La leucémie aiguë lymphoblastique est le cancer le plus fréquent chez l’enfant. Elle fait l’objet d’un traitement adapté au risque et est curable dans la majorité des cas. De nouveaux médicaments innovants, comme les immunothérapies, sont en cours d’essais cliniques.
La leucémie aiguë lymphoblastique (LAL) est le cancer le plus fréquent chez l’enfant, avec une proportion d’environ 30% et 3,3 nouveaux cas pour 100 000 habitants de moins de 15 ans. Le pic d’âge se situe autour de cinq ans. En Suisse, environ 50 à 60 enfants sont atteints de LAL chaque année. Le sous-type le plus fréquent sur le plan immunologique chez l’enfant est la LAL précurseur B, qui se développe à partir de cellules immatures de la série B du système lymphatique. Les ALL de lymphopoïèse T sont plus rares. Une forme particulière est la leucémie à cellules B matures, qui est due à une transformation maligne des cellules B matures et est considérée comme une manifestation leucémique d’un lymphome de Burkitt. La LAL est une maladie hétérogène qui se caractérise par une prolifération incontrôlée de cellules progénitrices lymphoïdes dans la moelle osseuse et le sang périphérique [1]. Elle est désormais considérée comme une maladie qui, bien que présentant souvent de grandes similitudes morphologiques, peut présenter des sous-entités très hétérogènes sur le plan cytogénétique ou moléculaire [2], ce qui est également corrélé à une réponse clinique hétérogène au traitement. Les techniques modernes de séquençage permettent de montrer l’énorme hétérogénéité clonale de cette maladie.
Causes
La cause de l’apparition de la leucémie n’est toujours pas claire. Les facteurs connus, mais peu fréquents, sont les radiations ionisantes et les syndromes congénitaux. Toutefois, cela explique moins de 10% de toutes les maladies. Les enfants atteints du syndrome de Down ont environ 20 fois plus de risques de développer une leucémie (LAL ou leucémie myéloblastique aiguë) au cours des cinq premières années de leur vie, par rapport aux enfants sains non atteints. Cependant, une myéloprolifération néonatale transitoire est encore plus fréquente chez ces enfants (3-10%) et peut parfois évoluer vers une leucémie. D’autres altérations congénitales rares présentant un risque accru de leucémie sont l’ataxie télangiectasique, le syndrome de Fanconi et d’autres syndromes associés à un déficit immunitaire ou à une fragilité chromosomique accrue. Le fait que l’ALL survienne plus fréquemment entre la deuxième et la cinquième année de vie, que l’on constate une augmentation de la maladie dans les pays industrialisés et que l’on ait observé par le passé des regroupements de cas, en particulier dans les régions de nouvelles agglomérations, a conduit à différentes hypothèses de leucémie associées à des infections [3,4].
Symptômes
La multiplication des blastes leucémiques dans la moelle osseuse entraîne le déplacement de l’hématopoïèse normale, ce qui explique les symptômes les plus courants tels que la pâleur et la fatigue dues à l’anémie ou la tendance aux saignements due aux thrombocytopénies. Les infiltrations provoquent souvent des douleurs osseuses diffuses et des arthropathies variables, qui se manifestent parfois chez les jeunes enfants par une réticence à bouger, voire un refus de marcher. En outre, il peut y avoir un gonflement des ganglions lymphatiques et des organomégalies.
Diagnostic
Dans le sang, on observe souvent des modifications d’au moins deux séries de cellules sanguines, le plus souvent une thrombocytopénie accompagnée d’une anémie. Le nombre de leucocytes peut être normal, abaissé ou augmenté. La morphologie de l’hémogramme fournit des indications diagnostiques importantes, le diagnostic final étant établi par ponction de moelle osseuse. Outre l’étude de la morphologie, elle permet de déterminer l’immunophénotype des blastes leucémiques par cytométrie de flux (FACS) et de réaliser une analyse chromosomique. L’immunophénotypage permet de déterminer le stade de développement du clone cellulaire correspondant. Le sous-type de leucémie le plus fréquent chez les enfants, appelé “common-ALL”, se caractérise par l’expression des marqueurs de cellules B CD10 et CD19. L’expression d’antigènes myéloïdes, qui n’a généralement pas de signification pronostique, peut être détectée dans la moitié des LAL. De nos jours, les analyses cytogénétiques et moléculaires occupent une place de plus en plus importante. Il s’agit d’identifier les sous-groupes les plus importants, car ils ont des implications thérapeutiques. D’une part, on recherche aussi bien des modifications chromosomiques numériques telles que l’hyperdiploïdie ou l’hypodiploïdie que des modifications structurelles telles que des translocations, par exemple t(12;21) (fusion des gènes ETV6/RUNX1) ou t(9;22) (fusion de BCR/ABL1), des réarrangements MLL (MLL 11q23) et d’autres modifications.
Classiquement, la détection de ces modifications se fait par cytogénétique conventionnelle (G-banding) et/ou par hybridation in situ fluorescente (FISH) dans les cellules leucémiques. Dans le cadre du diagnostic évolutif visant à évaluer la réponse de la leucémie au traitement, la mesure de la maladie résiduelle minimale (“minimal residual disease”, MRD) à partir de la moelle osseuse s’est établie au cours des dernières années. La réponse au traitement s’est désormais révélée être l’un des paramètres pronostiques les plus importants. Pour le diagnostic de l’évolution, on utilise aujourd’hui essentiellement deux méthodes qui se complètent dans la pratique clinique quotidienne. La méthode la plus sensible est le suivi des réarrangements des immunoglobulines et des récepteurs des lymphocytes T. Les réarrangements clonaux spécifiques à la leucémie sont initialement recherchés et suivis par PCR quantitative à des moments précis du traitement. La limite de détection ainsi atteinte est d’environ une cellule leucémique sur 100 000 cellules normales de la moelle osseuse. Une technique de mesure de la MRD moins sensible d’environ un niveau log repose sur le suivi du phénotype immunitaire associé à la leucémie par FACS. Une sensibilité de 0,001% peut être atteinte [5].
Traitement
Dans les années 70, moins de 30% des enfants survivaient à la maladie, alors qu’aujourd’hui, près de 85% des patients peuvent être guéris à long terme (Fig. 1). Les progrès réalisés en matière de survie au cours des dix à quinze dernières années sont principalement dus à une meilleure stratification des risques et, par conséquent, à un traitement adapté à ces risques. Le traitement de première ligne actuel de la LAL consiste essentiellement en l’association de corticostéroïdes, d’une déplétion en acides aminés ou en substrats (asparaginase, méthotrexate), de substances alkylantes, d’antimétabolites, de bloqueurs de métaphases classiques et d’anthracyclines [6]. Les nouvelles substances, appelées thérapies “ciblées”, n’ont été utilisées que de manière très limitée dans le traitement de la LAL pédiatrique, à quelques exceptions près, comme les inhibiteurs de tyrosine kinase dans la LAL à chromosome Philadelphie positif. Plus récemment, de nouveaux sous-groupes rares d’ALL précurseurs B ont été identifiés, appelés “Philadelphia like” (ou “BCR-ABL like”), qui présentent d’une part un risque de récidive nettement plus élevé, mais qui pourraient d’autre part être des candidats potentiels pour des thérapies ciblées [7]. Cependant, pour la plupart des nouveaux critères génétiques à haut risque, il a également été démontré que leur influence varie en fonction de la réponse thérapeutique mesurable.
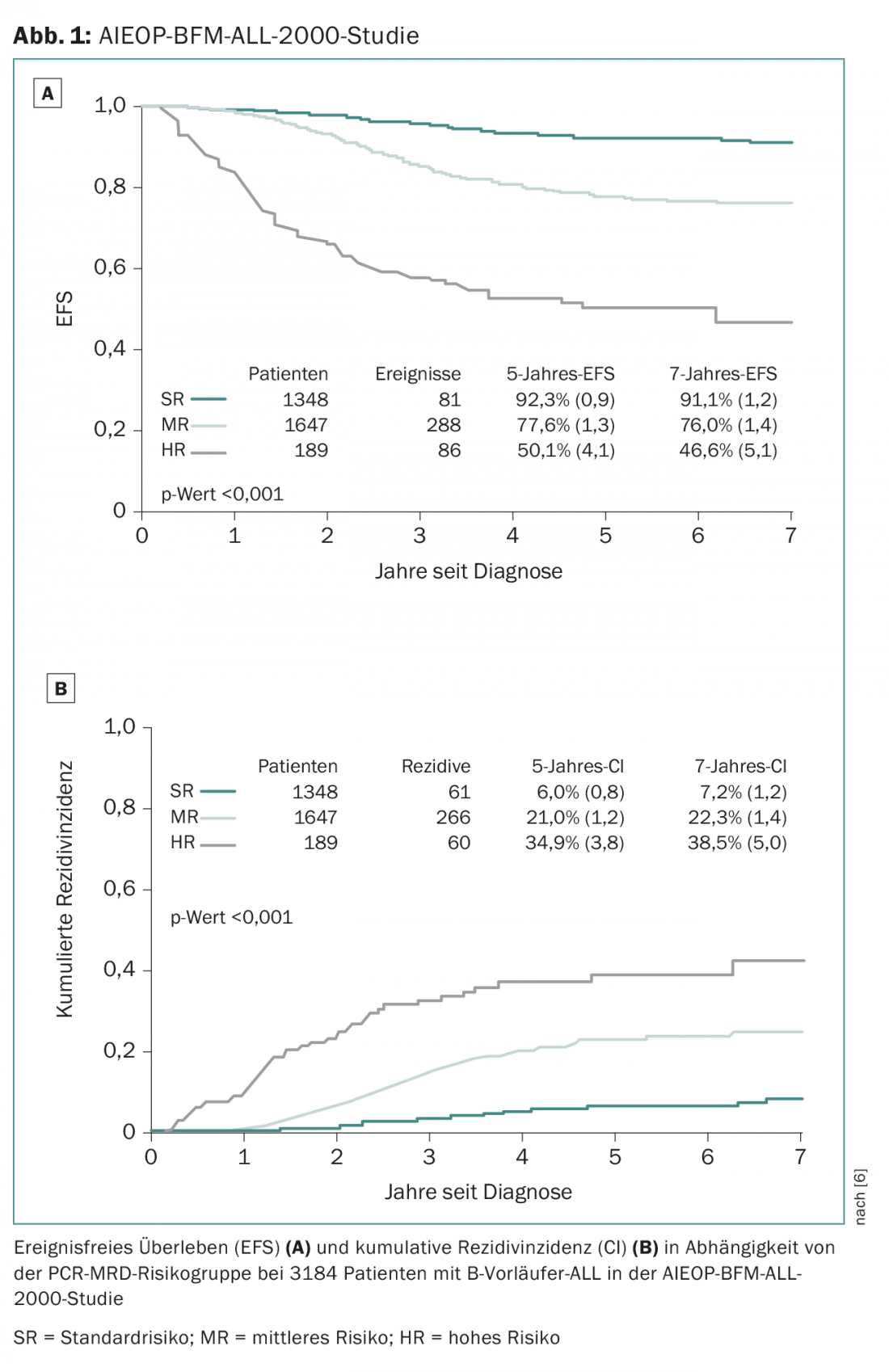
La plupart des centres d’oncologie pédiatrique suisses traitent leurs patients dans le cadre d’études du groupe d’étude ALL-BFM, une association de centres d’oncologie pédiatrique allemands, autrichiens et suisses qui, depuis 1976, a largement contribué à l’amélioration du traitement de la LAL dans le cadre de nombreux essais thérapeutiques randomisés à grande échelle. Alors que les études ALL-BFM précédentes ont permis de réaliser la plupart des progrès en adaptant la classification des groupes à risque et en individualisant le traitement, l’étude de suivi AIEOP-BFM ALL 2017, actuellement en cours de planification, utilisera pour la première fois de nouveaux médicaments prometteurs. Les médicaments classiques mentionnés pour le traitement de la LAL constituent la “colonne vertébrale” du traitement. En outre, les nouveaux médicaments innovants sont évalués de manière randomisée pour leur bénéfice potentiel. L’étude AIEOP-BFM-2017 prévue définit de nouveaux groupes cytogénétiques à haut risque (aperçu 1) qui auront accès à de nouvelles approches thérapeutiques innovantes. L’un de ces nouveaux sous-groupes est défini par la présence d’une délétion IKZF1 associée à une délétion CDKN2A, CDKN2B, PAX5 ou PAR1 en l’absence de délétion ERG et est appelé IKZF1plus. Dans des études antérieures, environ 10 à 15% des patients ont été identifiés comme IKZF1plus et ces patients présentaient un taux de récidive significativement plus élevé que les patients IKZF1plus-négatifs [8]. L’un de ces nouveaux médicaments, qui sera utilisé dans un petit groupe de groupes à haut risque et au pronostic particulièrement défavorable, est le blinatumomab, un anticorps bispécifique contre les cellules T (BiTE) qui cible simultanément le récepteur CD3 des cellules T et la protéine de surface CD19 des cellules B [9]. Le blinatumomab devrait combiner deux effets potentiels : Réduire les toxicités aiguës et à long terme en économisant la chimiothérapie conventionnelle et traiter plus efficacement les patients qui n’ont jusqu’à présent répondu que de manière insatisfaisante à la thérapie à haut risque.

Le bortézomib, un inhibiteur du protéasome, est un autre nouveau médicament doté d’un nouveau mode d’action dans le traitement de première ligne de la LAL. Étant donné que les tentatives précédentes d’intensification tardive du traitement chez les patients à haut risque n’ont pas été couronnées de succès et en raison des toxicités déjà élevées des traitements à haut risque (HR), le bortézomib sera utilisé de manière randomisée dès la phase post-réinduction précoce chez les patients HR dans l’étude à venir.
Système nerveux central (SNC)
La prévention d’une récidive du SNC se fait aujourd’hui majoritairement par voie médicamenteuse, d’une part par des injections intrathécales de méthotrexate, d’autre part par l’administration de cytostatiques à action systémique qui s’infiltrent dans le cerveau (par ex. méthotrexate à haute dose). Ainsi, l’ancienne radiothérapie du SNC, qui a certes permis une réduction spectaculaire des récidives du SNC, mais qui était associée à des séquelles non négligeables, a pu être limitée à des situations à risque très particulières [10,11].
Transplantation de cellules souches
Les résultats du traitement primaire et des protocoles de récidive des leucémies se sont nettement améliorés au fil du temps, ce qui a conduit à une adaptation continue des indications des thérapies à haute dose avec réinjection de cellules souches. L’indication actuelle d’une transplantation de cellules souches (TCS) dans le cadre du traitement primaire est réservée à certains sous-groupes cytogénétiques de mauvais pronostic tels que t(9;22), les hypodiploïdies avec moins de 44 chromosomes dans les blastes ainsi que IKZF1plus en combinaison avec une réponse thérapeutique insuffisante (MRD) au fil du temps [12]. L’expérience du groupe BFM a montré que le succès du traitement des récidives dépend du moment de la survenue de la récidive, du type d’atteinte de la leucémie et du sous-type de leucémie [13]. Mais là encore, il a été démontré que la réponse au traitement après une nouvelle induction thérapeutique, et donc la dynamique de la diminution de la maladie résiduelle minimale, revêt une importance pronostique particulière et que d’autres éléments thérapeutiques, tels que le recours à une TSE, peuvent être orientés en conséquence [14].
Nouvelles thérapies
A quelques exceptions près (clofarabine, nelarabine, imatinib), il n’y a pas eu de nouvelles autorisations de mise sur le marché pour les LAL pédiatriques au cours des 10 à 15 dernières années. Cependant, plusieurs approches thérapeutiques intéressantes sont actuellement en cours d’évaluation clinique dans le cadre d’essais de phase I-III. Outre le blinatumomab susmentionné, il s’agit notamment de cellules T chimériques du récepteur de l’antigène (CAR), qui ont déjà été utilisées avec succès pour traiter des récidives de LAL CD19 positives. Il s’agit là encore d’une immunothérapie qui exploite le potentiel des lymphocytes T cytotoxiques autologues, capables de reconnaître et de détruire les cellules de la lignée des lymphocytes B. D’autres immunothérapies prometteuses, parfois couplées à des cytostatiques, sont actuellement en phase I/II d’essais. Outre les immunothérapies, les thérapies ciblées après test préalable in vitro dans des modèles de xénogreffes ou de lignées cellulaires, ou les inhibiteurs spécifiques contre les gènes de fusion détectés par cytogénétique, constituent des options thérapeutiques intéressantes et prometteuses.
Messages Take-Home
- La leucémie lymphoblastique aiguë, le cancer le plus fréquent chez l’enfant, est traitée de manière adaptée au risque et peut être guérie dans la majorité des cas.
- La détermination de la maladie résiduelle minimale après induction du traitement est l’un des facteurs pronostiques les plus importants, en plus des marqueurs biologiques tels que le sous-type leucémique et les modifications cytogénétiques et moléculaires des blastes leucémiques.
- Les développements actuels visent à traiter plus efficacement et de manière plus ciblée les sous-types de leucémie jusqu’à présent résistants et à réduire la toxicité du traitement.
- De nouveaux médicaments innovants, tels que les immunothérapies et les approches thérapeutiques individualisées, sont en cours d’essais cliniques.
Littérature :
- Jabbour E, et al : New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer 2015 ; 121(15) : 2517-2528.
- Pui CH, et al : Biologie, stratification des risques, et traitement des leucémies aiguës pédiatriques : une mise à jour. J Clin Oncol 2011 ; 29(5) : 551-565.
- Kinlen L, et al : Infections et facteurs immunitaires dans le cancer : le rôle de l’épidémiologie. Oncogene 2004 ; 23 : 60-75.
- Greaves M, et al : Infection, réponses immunitaires et l’étiologie de la leucémie infantile. Nat Rev Cancer 2006 ; 6(3) : 193-203.
- Campano D, et al : Minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Curr Opin Hematol 2012 ; 19 : 313-318.
- Conter V, et al : La réponse moléculaire au traitement redéfinit tous les facteurs pronostiques chez les enfants et les adolescents atteints de leucémie lymphoblastique aiguë à précurseurs de cellules B : résultats chez 3184 patients de l’étude AIEOP-BFM ALL 2000. Blood 2010 ; 115(16) : 3206-3214.
- Loh ML, et al : Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia : a report from the Children’s Oncology Group TARGET Project. Blood 2013 ; 121(3) : 485-488.
- Hinze L, et al : Impact pronostique des délétions d’IKZF1 en association avec les pulses de vincristine-dexaméthasone pendant le traitement d’entretien de la leucémie aiguë lymphoblastique de l’enfant sur le trial ALL-BFM 95. Leucémie 2017 ; 31 : 1840-1842.
- Brentjens RJ, et al : Sécurité et persistance des cellules T autologues ciblées par CD19 transférées de manière adoptive chez les patients atteints de leucémie à cellules B en rechute ou réfractaire à la chimiothérapie. Blood 2011 ; 118(18) : 4817-4828.
- Möricke A, et al : Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival : treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 2008 ; 111(9) : 4477-4489.
- Kamps WA, et al : BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients : results of DCLSG protocol ALL-8 (1991-1996). Leucémie 2002 ; 16(6) : 1099-1111.
- Balduzzi A, et al : Chimiothérapie versus transplantation allogénique pour la leucémie lymphoblastique aiguë à très haut risque de l’enfant en première rémission complète : comparaison par randomisation génétique dans une étude prospective internationale. Lancet 2005 ; 366 : 635-642.
- Tallen G, et al : Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy : results of trial ALL-REZ BFM 90. J Clin Oncol 2010 ; 28 : 2339-2347.
- Eckert C, et al : Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia – Long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 2013 Apr ; 49(6) : 1346-1355.
InFo ONKOLOGIE & HÄMATOLOGIE 2017 ; 5(5) : 30-33









