Les signes cutanés pathognomoniques de la dermatomyosite comprennent l’érythème héliotrope, les papules de Gottron et le signe de Gottron. La détermination des auto-anticorps spécifiques à la myosite est de plus en plus importante pour le diagnostic. En ce qui concerne les possibilités de traitement, les immunoglobulines intraveineuses ont récemment été officiellement autorisées en Suisse. Les corticostéroïdes et les traitements de fond conventionnels sont toujours considérés comme la norme thérapeutique. Les produits biologiques et les “petites molécules” sont utilisés en off-label.
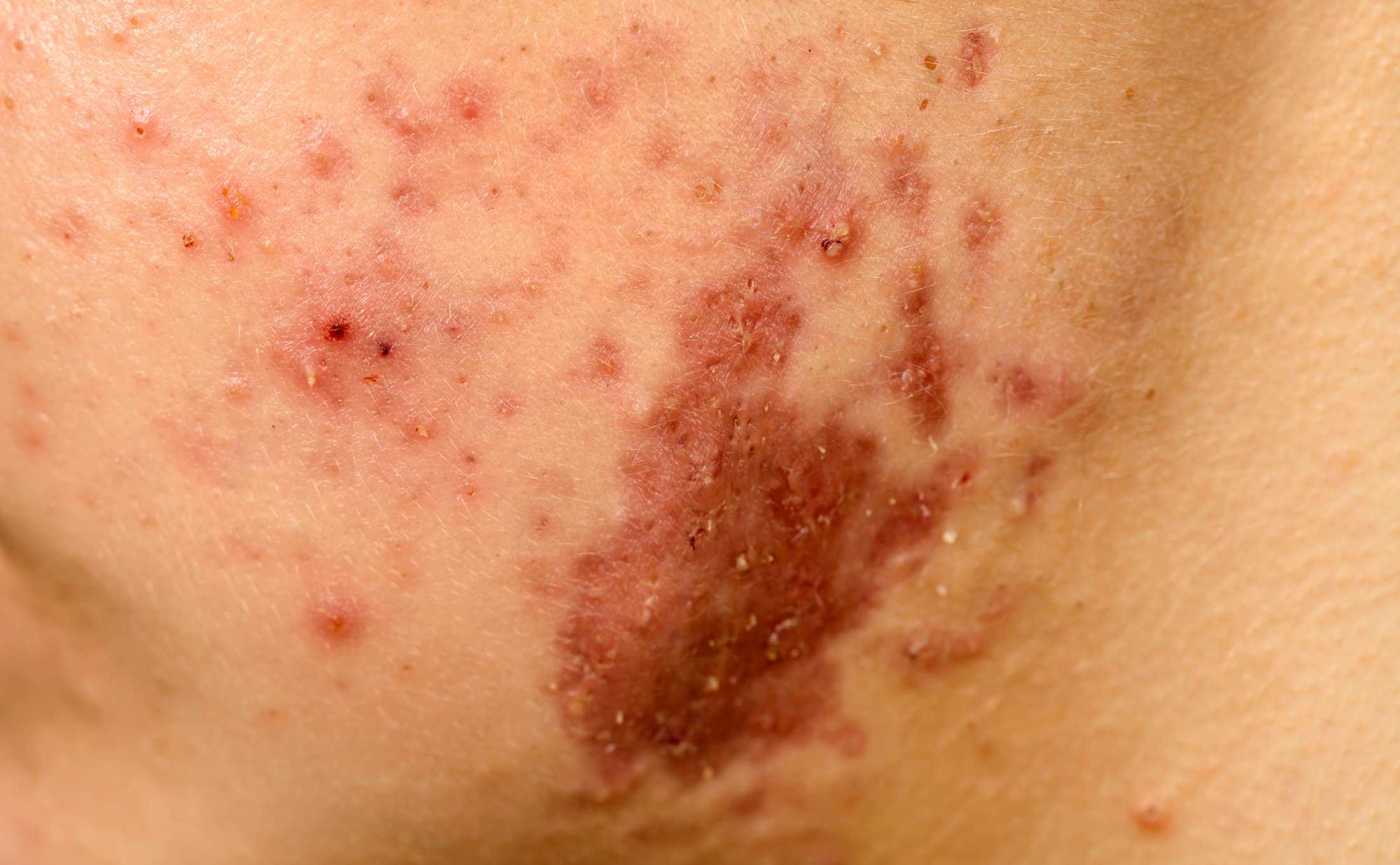
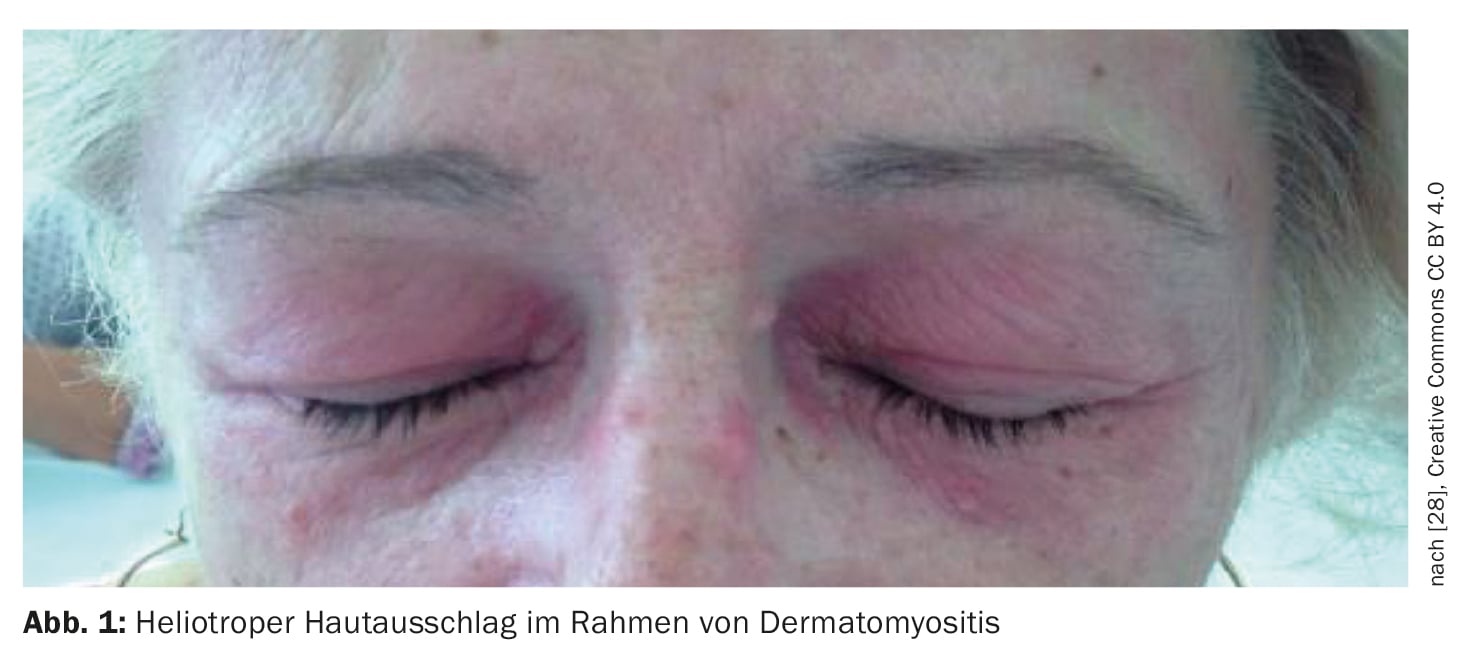
La dermatomyosite (DM) est une maladie auto-immune appartenant au groupe des myosites inflammatoires idiopathiques (encadré) , qui peut débuter dès l’enfance mais aussi se manifester à l’âge adulte avancé [1–3]. Les taux d’incidence les plus élevés sont documentés entre 5 et 14 ans (DM juvénile) et entre 45 et 50 ans. Les femmes sont environ deux fois plus touchées que les hommes, et il n’y a pas d’association familiale connue [4]. Dans le cadre de l’atteinte musculaire, il se produit une destruction musculaire inflammatoire qui peut entraîner une faiblesse musculaire plus ou moins prononcée, surtout au niveau des muscles de la ceinture scapulaire et pelvienne proches du tronc. Les signes classiques sur la peau comprennent l’éruption héliotrope (figure 1), l’érythème des joues, le signe de Gottron (figure 2) et les modifications du lit de l’ongle. En plus de la faiblesse musculaire et des changements typiques de la peau, il y a parfois une implication cardiaque et pulmonaire. Le professeur Britta Maurer, directrice de clinique et médecin-chef, Clinique de rhumatologie, Hôpital de l’Île, Berne, a donné un aperçu actuel du diagnostic et du traitement de la dermatomyosite [5].

Auto-anticorps spécifiques à la myosite, IRM et biopsie si nécessaire
Dans la pathogenèse de la dermatomyosite, une microangiopathie médiée par le complément avec des infiltrats lymphocytaires périvasculaires de la peau et des muscles jouent un rôle décisif [6]. Dans une classification des sociétés savantes européennes et américaines (EULAR, ACR), les paramètres d’examen clinique et de laboratoire sont utilisés pour calculer la probabilité de diagnostic d’un DM (encadré) [2,3,7].

www.imm.ki.se/biostatistics/calculators/iim
Les paramètres de myolyse, à savoir la créatine kinase (CK), la myoglobine et la lactate déshydrogénase (LDH), sont souvent peu élevés, voire normaux, dans la DM [4]. La détection de certains auto-anticorps est associée à différentes expressions phénotypiques de la dermatomyosite (tableau 1). Cependant, dans environ 30% des cas de DM, aucun auto-anticorps n’est détecté dans le sérum des patients [8].
L’imagerie par résonance magnétique (IRM) peut être utilisée pour visualiser les infiltrats musculaires inflammatoires. Les biopsies sont particulièrement utiles dans les cas peu clairs. Une biopsie musculaire permet de détecter des infiltrats inflammatoires, périvasculaires et/ou périfasciculaires. Les résultats histopathologiques des lésions cutanées de la DM comprennent la dégénérescence vacuolaire des kératinocytes basaux, des infiltrats inflammatoires lymphocytaires autour des vaisseaux sanguins de la peau et des dépôts de mucine interstitielle [9].
La recherche de tumeurs est recommandée pour toutes les formes de DM, car il existe un risque statistiquement accru (aperçu 1) [7]. La plupart des recherches de tumeurs sont effectuées au cours de la première année après le diagnostic de DM et jusqu’à trois ans après le diagnostic, car c’est la période où les manifestations tumorales sont les plus fréquentes.

Corticostéroïdes en phase d’induction – stratégie d’économie de stéroïdes au cours de l’évolution
Les glucocorticostéroïdes sont le traitement de première intention de la DM (figure 3) [8]. On commence par 1 mg/kg PC par jour pendant la phase aiguë jusqu’à l’amélioration clinique, puis on réduit lentement la dose. La plupart des patients répondent bien dans un premier temps, mais pour économiser les stéroïdes, il convient d’administrer un immunosuppresseur supplémentaire au plus tard après 6 mois ; un traitement combiné peut être envisagé dès le départ, en particulier dans les formes graves (figure 3).

Les glucocorticostéroïdes plus l’azathrioprine sont la combinaison la plus courante dans le traitement de la DM [8]. L’azathioprine doit être administrée à une dose de 1 à 3 mg/kg PC, en particulier dans les formes graves, par exemple en cas de faiblesse généralisée, d’atteinte des muscles respiratoires ou de déglutition, et ce dès le début, mais elle présente une latence connue de 3 à 6 mois avant d’agir.
Le méthotrexate (MTX) est au moins équivalent à l’AZA dans la DM avec une atteinte cutanée marquée et est préféré à l’AZA dans la DMJ lorsque la fonction rénale est normale. Le MTX est un antagoniste de l’acide folique et agit plus rapidement que l’AZA à une dose de 15-25 mg/semaine, mais il est également classé dans une classe de toxicité plus élevée. Une pneumonie est parfois observée comme effet secondaire.
En cas d’échec de l’AZA ou d’atteinte hépatique toxique, le mycophénolate mofétil (MMF, 2 g/d) peut être utilisé comme alternative. La ligne directrice actuelle [8] fait référence à plusieurs rapports de cas qui soutiennent les avantages de cette option de traitement [13–15]. Le MMF bloque sélectivement la synthèse des purines dans les lymphocytes et inhibe ainsi leur prolifération. Les principaux effets secondaires sont une diarrhée chronique, une anémie hémolytique et des œdèmes.
L’IVIG désormais officiellement approuvée par Swissmedic
Chez les patients qui ne répondent pas suffisamment aux glucocorticoïdes/AZA, il est utile d’essayer un traitement par immunoglobulines intraveineuses (figure 3). Il existe des preuves d’efficacité convaincantes pour les IgIV (Octagam®), qui ont été confirmées dans l’étude d’homologation de phase III en double aveugle et contrôlée par placebo ProDERM [18,19]. En Suisse, Octagam® a reçu une extension d’indication de Swissmedic en février 2023 pour le traitement de la DM chez l’adulte [30].
En cas d’évolution réfractaire, la ligne directrice recommande le rituximab
Le rituximab (RTX), un anticorps anti-CD20 utilisé en off-label dans la DM en Suisse, s’est révélé prometteur dans les cas réfractaires (figure 3) [16]. En ce qui concerne la posologie, la ligne directrice recommande d’utiliser le “schéma immunologique” de 2× 1000 mg i.v. à 14 jours d’intervalle, avec éventuellement une nouvelle administration après environ 6 à 9 mois si cela semble nécessaire au vu de l’évolution clinique. Les patients du registre GRAID-2 ont reçu en moyenne 3,09 perfusions [16,17]. Ces patients n’ont pas présenté de problèmes de sécurité particuliers et la plupart d’entre eux ont montré une bonne tolérance. Les infections sont les principaux effets secondaires indésirables d’un traitement par rituximab [16,17].
Si ces stratégies thérapeutiques ne donnent pas de résultats : ce qu’il y a d’autre à faire
Dans une petite étude, l’abatacept, qui fait partie des médicaments biologiques, a montré des résultats prometteurs en termes de réduction de l’activité de la maladie chez des patients adultes atteints de dermatomyosite réfractaire [21]. Après six mois de traitement par abatacept, les biopsies musculaires ont montré une augmentation des cellules anti-inflammatoires Fork-Head-Box-P3 (FoxP3) + T régulatrices (Treg), ce qui indique une régénération musculaire et une réponse au traitement [21]. Tang et al. ont étudié les effets de l’abatacept chez les patients atteints de myosite en utilisant un ensemble de données de l’étude ARTEMIS et ont constaté que le rapport CD4/CD8 dans la circulation sanguine au moment de la maladie active pourrait être un prédicteur de l’efficacité du traitement [22]. L’examen de l’efficacité et de la sécurité de l’abatacept dans la dermatomyosite fait l’objet de plusieurs études récentes [16].
Plusieurs cas de traitement par tofacitinib, un inhibiteur de JAK, ont été rapportés, notamment ceux de deux patients atteints de dermatomyosite avec une calcinose et une pneumopathie interstitielle, qui ont été traités par tofacitinib sur une période de 28 semaines et ont montré une très bonne réponse. Aucune nouvelle calcification n’est apparue chez les deux patients, le traitement par tofacitinib a été bien toléré et aucun problème de sécurité majeur n’est survenu [23]. Dans un autre cas rapporté, un patient adulte atteint de dermatomyosite hypomyopathique positive aux anticorps anti-MDA-5 et anti-Ro52 avec atteinte pulmonaire interstitielle a montré une réponse significative au tofacitinib, qui s’est traduite par une amélioration des performances physiques, ainsi que de l’état de la peau et de la pneumopathie interstitielle. Le traitement a été bien toléré [24]. L’utilisation du tofacitinib est également discutée pour le traitement de la dermatomyosite réfractaire anti-NXP2 et anti-TIF1γ [25].
Des séries de cas indiquent également que l’inhibiteur oral de la PDE-4, l’aprémilast, pourrait être utile chez les patients présentant des symptômes récurrents de DM, en complément d’autres médicaments immunomodulateurs [26]. Chez trois patients, l’aprémilast 30 mg (2×/j) en add-on a entraîné une nette amélioration et des effets d’économie de stéroïdes. Le mécanisme d’action exact de l’aprémilast dans la DM n’est pas connu. On pense que l’influence sur la réponse Th1 et Th2 est impliquée [26].
Outre l’abatacept, le tofacitinib et l’aprémilast, l’ustékinumab et le cyclophosphamide font partie des molécules utilisées hors étiquette dans les cas autrement réfractaires et font l’objet d’essais cliniques en cours.
Congrès : Allergy and Immunology-Update (SSAI)
Littérature :
- Dressler F, Maurer B : Dermatomyosite et dermatomyosite juvénile. Z Rheumatol 2022. https://doi.org/10.1007/s00393-022-01205-5
- Tomaras S, Kekow J, Feist E : Myopathies inflammatoires idiopathiques : actualités sur le diagnostic et la classification. Akt Rheumatol 2021 ; 46 : 361-372.
- Bottai M, et al : International Myositis Classification Criteria Project consortium, the Euromyositis register and the Juvenile Dermatomyositis Cohort Biomarker Study and Repository (JDRG) (UK and Ireland). Critères de classification EULAR/ACR pour les myopathies inflammatoires idiopathiques adultes et juvéniles et leurs principaux sous-groupes : un rapport méthodologique. RMD Open 2017 Nov 14;3(2) : e000507.
- Deutsche Gesellschaft für Muskelkranke e.V. : DGM-Handbuch Myositis, 2020, chapitre 3a. Dermatomyosite (DM), Prof Dr Eugen Feist, Prof Dr Cord Sunderkötter. Guide du patient myosite2020web.pdf
- “Dermatomyosite – Current Diagnostic Approaches and Therapeutic Strategies”, Prof. Dr. med. Britta Maurer, Allergy and Immunology-Update (SGAI), 27-29.01.23.
- Stuhlmüller B, et al : New aspects on the pathogenesis of myositis. Z Rheumatol 2013 ; 72 : 209-219.
- Schlecht N, et al : Mise à jour sur la dermatomyosite chez l’adulte. J Dtsch Dermatol Ges 2020 ; 18(9) : 995-1013.
- “Myositisyndrome”, ligne directrice s2k, Commission des lignes directrices de la Société allemande de neurologie (éd.), révision complète : 28.04.2022.
- Okiyama N, Fujimoto M : Manifestations cutanées de la dermatomyosite caractérisées par des auto-anticorps spécifiques de la myosite. F1000Res 2019 Nov 21 ; 8 : F1000 Faculty Rev-1951. doi : 10.12688/f1000research.20646.1.
- Sell S : Dermatomyositis im multiregionalen Vergleich, thèse de doctorat, Faculté de médecine de l’Université Friedrich-Alexander, Erlangen-Nuremberg, 2021.
- Hill CL, et al : Fréquence de types spécifiques de cancer dans la dermatomyosite et la polymyosite : une étude basée sur la population. Lancet 2001 : 357(9250) : 96-100.
- Stockton D, Doherty V, Brewster D : Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications : a Scottish population-based cohort study. British journal of cancer 2001 ; 85(1) : 41-45.
- Majithia V, Harisdangkul V : Mycophénolate mofetil (CellCept) : un traitement alternatif pour la myopathie inflammatoire auto-immune. Rhumatologie (Oxford) 2005 ; 44(3) : 386-389.
- Schneider-Gold C HH, Gold R. Mycophénolate mofetil et tacrolimus : nouvelles options thérapeutiques dans les maladies neuro-immunologiques. Nerf musculaire 2006 ; 34 : 284-291.
- Chaudhry V, et al. : Mycophénolate mofetil : un immunosuppresseur sûr et prometteur dans les maladies neuromusculaires. Neurology 2001 ; 56(1) : 94-96.
- Patil A, et al : Traitement de la dermatomyosite adulte et juvénile. J Cosmet Dermatol 2023 ; 22(2) : 395-401.
- 17. Fiehn C, et al : Rituximab pour le traitement des poly- et dermatomyosites : résultats du registre GRAID-2. Z Rheumatol 2018 ; 77 : 40-45.
- Aggarwal R, et al. Efficacité et sécurité de l’IVIg (Octagam 10%) chez les patients atteints de dermatomyosite active. Résultats d’un essai de phase III randomisé, en double aveugle et contrôlé par placebo (étude ProDERM) [abstract]. Arthritis Rheumatol 2020 ; 72.
- Dalakas MC, et al : A controlled trial of high-dose intravenous immunoglobulin infusions as treatment for dermatomyositis. N Engl J Med 1993 ; 329(27) : 1993-2000.
- Stringer E, Feldman BM : Advances in the treatment of juvenile dermatomyositis. Curr Opin Rheumatol. 2006 ; 18(5) : 503-506.
- Tjarnlund A, et al : L’abatacept dans le traitement de la dermatomyosite et de la polymyosite de l’adulte : un essai randomisé de phase IIb de traitement à début différé. Ann Rheum Dis 2018 ; 17 : 55-62.
- Tang Q, et al. Effet du traitement par CTLA4-Ig (abatacept) sur les cellules T et les cellules B dans le sang périphérique de patients atteints de polymyosite et de dermatomyosite. Scand J Immunol 2019 ; 89:e12732.
- 23 Wendel S, et al : Successful treatment of extensive calcifications and acute pulmonary involvement in dermatomyositis with the Janus-kinase inhibitor tofacitinib – a report of two cases. J Autoimmun 2019 ; 100 : 131-136.
- Hornig J, et al : Réponse de la dermatomyosite avec implication pulmonaire au traitement par l’inhibiteur de Janus kinase. Z Rheumatol 2018 ; 77 : 952-957.
- Navarro-Navarro I, et al : Treatment of refractory anti-NXP2 and anti-TIF1γ dermatomyositis with tofacitinib. J Dtsch Dermatol Ges 2020 ; 19 : 443-447.
- Bitar C, et al : Apremilast as a potential treatment for moderate to severe dermatomyositis : a retrospective study of 3 patients. JAAD Case Rep 2019 ; 5 : 191-194.
- Charlton D, et al : Dermatomyosite cutanée réfractaire avec prurit sévère du cuir chevelu sensible à l’apremilast. J Clin Rheumatol 2019 ; 27:S561-S562.
- 28. Bobirca A, et al. : Dermatomyosite amyopathique anti-MDA5 – Un défi diagnostique et thérapeutique. Life 2022 ; 12(8) : 1108. https://doi.org/10.3390/life12081108
- Żychowska M, Reich A : Dermoscopy and Trichoscopy in Dermatomyositis-A Cross-Sectional Study. Journal of Clinical Medicine 2022 ; 11(2):375. https://doi.org/10.3390/jcm11020375.
- Informations sur les médicaments, www.swissmedicinfo.ch/ViewMonographie,(dernière consultation 03.03.2023).
DERMATOLOGIE PRAXIS 2023 ; 33(2) : 47-49 (publié le 20.4.23, ahead of print)