Cet article donne un aperçu des différentes formes de dermatoses bulleuses auto-immunes et un résumé des recommandations thérapeutiques et des options de traitement actuellement en vigueur.
Les maladies auto-immunes vésicantes sont généralement des maladies graves de la peau – et souvent aussi des muqueuses. Elles se caractérisent par l’apparition d’auto-anticorps IgG ou IgA contre les protéines structurelles de la peau. Ces protéines structurelles sont très importantes pour l’adhésion cellulaire des kératinocytes (maladies pemphigus) ou pour l’adhésion de l’épiderme au derme (maladies pemphigus, épidermolyse bulleuse acquise, dermatite herpétiforme).
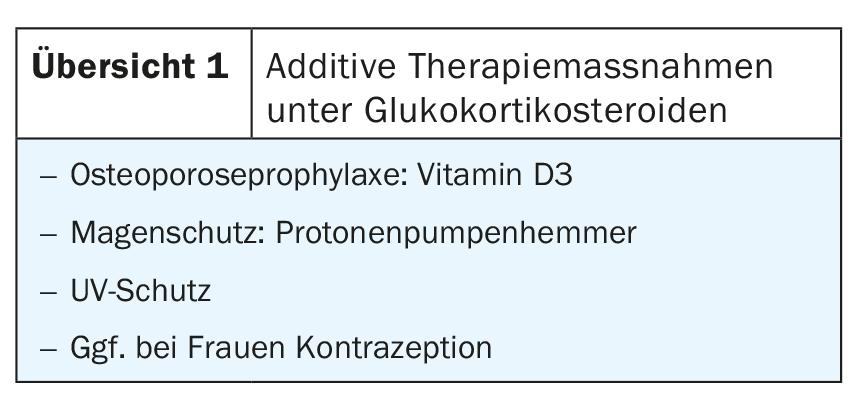
La formation de bulles est donc intraépidermique dans les pemphigus et sous-épidermique dans les autres maladies bulleuses auto-immunes. La grande hétérogénéité clinique et les différentes évolutions représentent également un défi thérapeutique pour les dermatologues. Les glucocorticostéroïdes locaux et oraux constituent le traitement de première intention, à l’exception de la dermatite herpétiforme. Sous traitement systémique par corticostéroïdes, des mesures thérapeutiques additives sont nécessaires (aperçu 1). Afin d’économiser les corticostéroïdes, ceux-ci sont associés à d’autres immunosuppresseurs au cours de l’évolution.
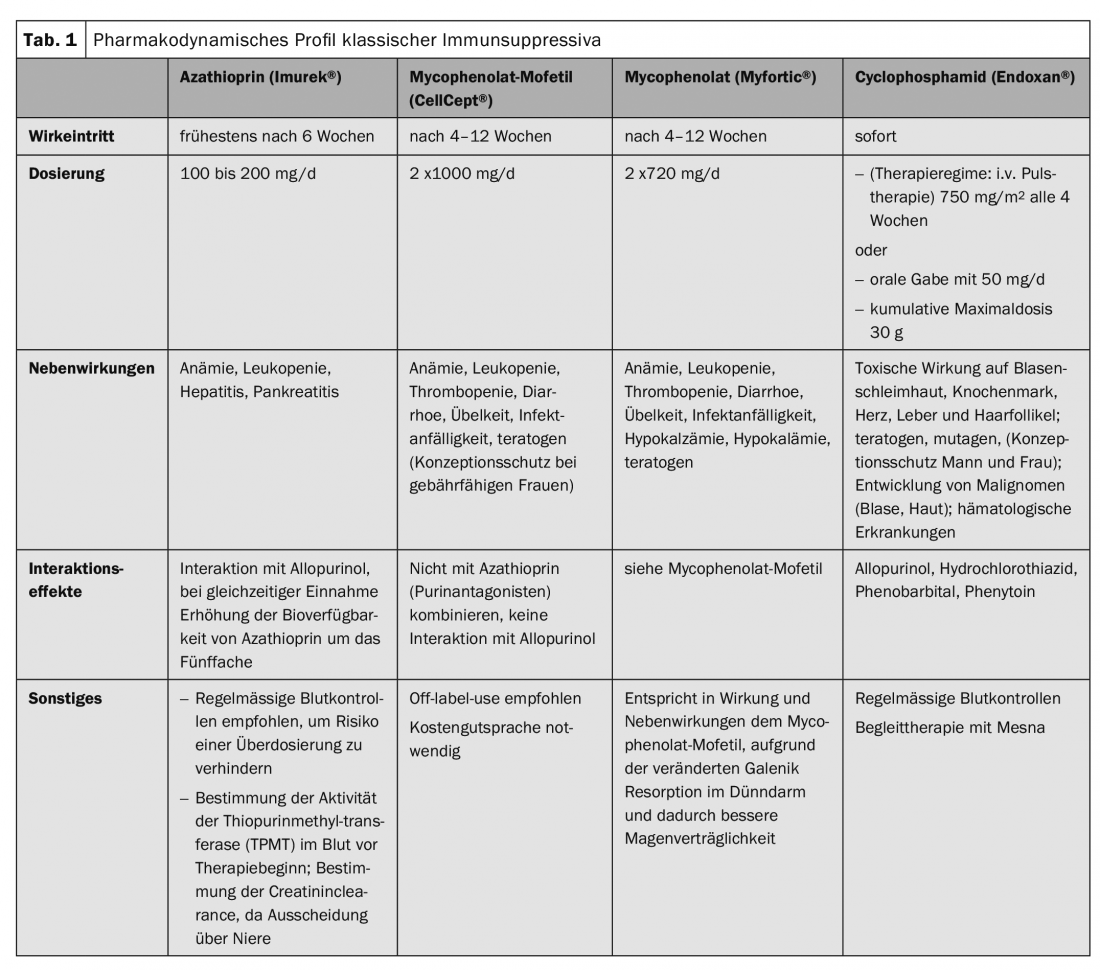
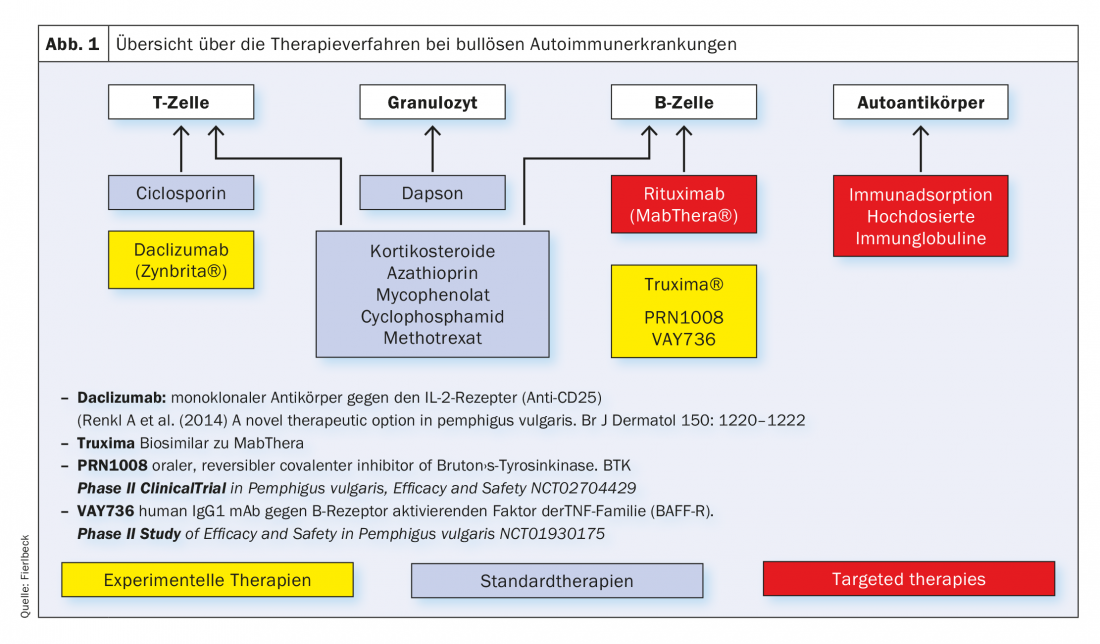
Au cours des dernières années, les immunosuppresseurs classiques (Tab. 1), qui influencent les processus métaboliques des cellules T et B au niveau cellulaire, complétées par des thérapies ciblées (“targeted therapies”) (Fig.1). Il s’agit essentiellement du rituximab (Tab.2), Procédure d’immunoadsorption (Tab. 3) et l’utilisation d’immunoglobulines à haute dose. (Tab.4). Le rituximab est un anticorps chimérique anti-CD20 destiné à la déplétion des cellules B périphériques qui produisent des auto-anticorps. (Tab.2).
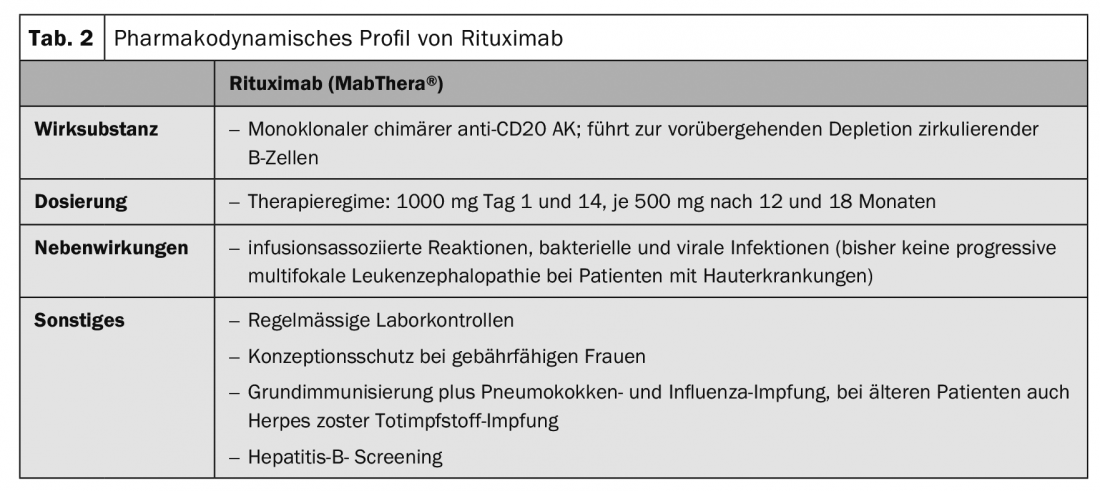
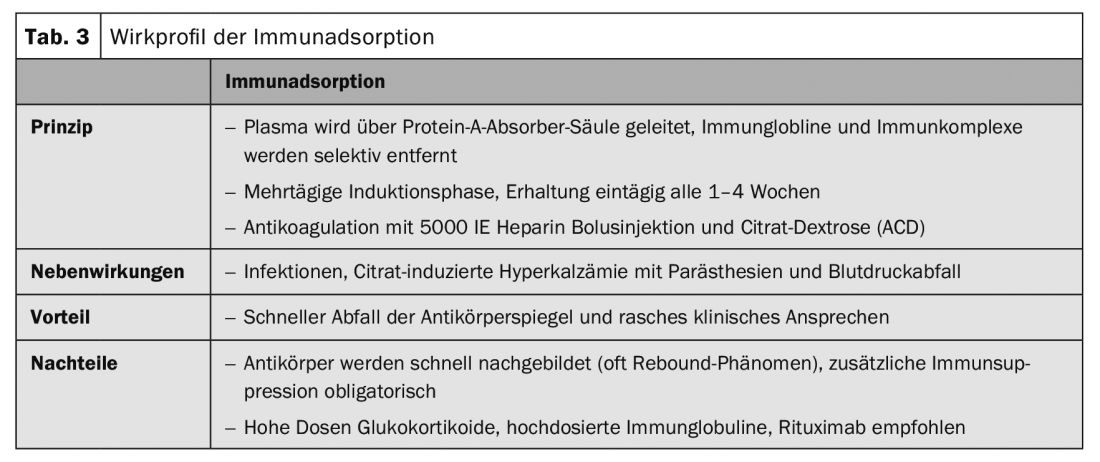
Les procédures d’immunoadsorption permettent d’éliminer sélectivement les autoanticorps et les complexes immuns du plasma à l’aide d’absorbants de haute affinité (tableau 3).
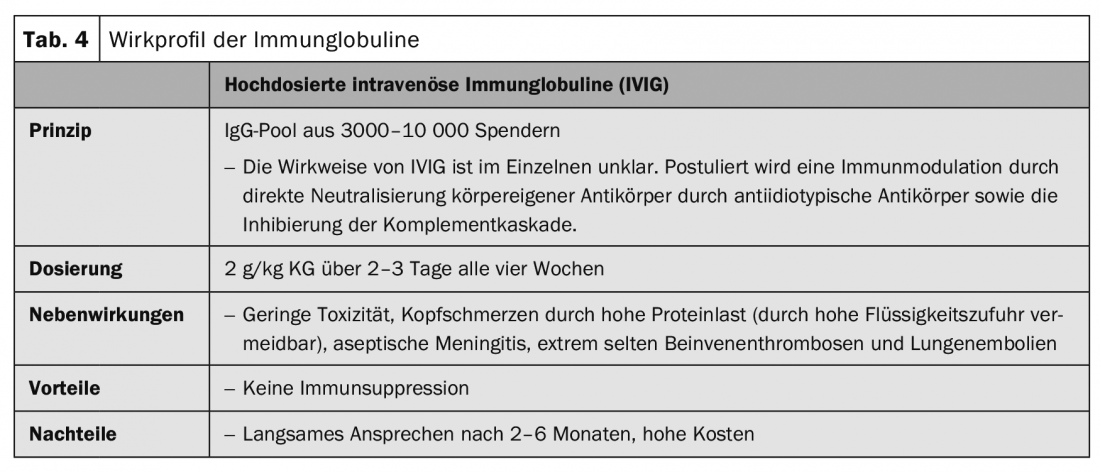
Les immunoglobulines administrées par voie intraveineuse à fortes doses visent à neutraliser les médiateurs inflammatoires et les auto-anticorps (tableau 4).
Sous traitement immunosuppresseur, il existe un risque accru de réactivation d’infections latentes, de sorte que les infections telles que le VIH, l’hépatite B, la tuberculose et les infections bactériennes chroniques doivent être exclues avant l’instauration du traitement. Le traitement peut entraîner des infections opportunistes à Candida ainsi que des infections herpétiques (herpès simplex, varicelle zoster). Un risque accru de maladies lymphoprolifératives et de tumeurs cutanées a été décrit dans le cadre d’un traitement immunosuppresseur à long terme. En raison de l’augmentation de la photocancérogénèse, une protection contre les UV et des inspections régulières de la peau sont nécessaires. Avant d’instaurer un traitement immunosuppresseur, il convient de vérifier le statut vaccinal du patient. Les vaccinations avec des vaccins vivants sous traitement immunosuppresseur sont contre-indiquées et les vaccinations avec des vaccins atténués peuvent réduire l’efficacité de la vaccination. Chez les patients âgés de plus de 50 ans, la vaccination contre le zona avec le vaccin contre le zona subunitaire est indiquée [1].
Maladies pemphigus
Ce terme générique regroupe le pemphigus vulgaire, le pemphigus foliacé et le pemphigus paranéoplasique. Les glucocorticostéroïdes systémiques sont au cœur du traitement. En fonction de la gravité de la maladie, les doses initiales correspondent à 1-2 mg/kg d’équivalent prednisolone par jour. Le traitement de consolidation dépend de l’activité de la maladie. Si aucune nouvelle bulle n’apparaît pendant 8 jours, la dose de stéroïdes est réduite de 25% toutes les 2 à 4 semaines. A partir de 30 mg d’équivalent prednisolone par jour, la réduction ultérieure est encore plus lente. L’objectif est d’atteindre le seuil de cushing (7,5 mg de prednisolone). Afin d’économiser les glucocorticostéroïdes, ceux-ci sont associés à d’autres immunosuppresseurs (tableau 1). La dose d’immunosuppresseur reste inchangée pendant toute la durée du traitement, à condition qu’il n’y ait pas d’effets secondaires.
L’immunosuppresseur adjuvant le plus courant est l ‘azathioprine, à une dose de 100-150 mg/j. En cas de diminution de l’activité enzymatique de la thiopurine méthyltransférase ou d’administration concomitante d’allopurinol, la dose doit être réduite. Des prises de sang sont nécessaires avant et pendant le traitement (tab. 1).
En cas d’effets secondaires sous azathioprine, le mycophénolate mofétil ou, en cas d’effets secondaires gastro-intestinaux, l’acide mycophénolique sont utilisés. Ces deux médicaments ont un effet d’épargne stéroïdienne et entraînent une rémission plus rapide (tab. 1).
En cas d’évolution très réfractaire, le cyclophosphamide peut être associé aux corticostéroïdes. Le cyclophosphamide est une substance alkylante, c’est-à-dire qui réticule l’ADN, avec des effets irréversibles sur la réserve ovarienne et la spermatogenèse, ce qui le rend obsolète chez les patients plus jeunes. En outre, un certain nombre d’effets secondaires toxiques et le développement de tumeurs malignes peuvent survenir (tableau 1). L’autorisation de mise sur le marché dans l’UE a donc été limitée en 2012 aux maladies auto-immunes mettant la vie en danger.
Des études cliniques prospectives, multicentriques et randomisées, avec ou sans corticostéroïdes, ont montré l’efficacité du rituximab (tableau 2) , avec des rémissions pouvant atteindre 80% à 24 mois. Les titres d’autoanticorps (antidesmogléine I et III) ont diminué lentement au cours des premières semaines après la perfusion et ont atteint leur niveau le plus bas après environ 180 jours. Une réponse clinique a été observée après 2-3 mois. Sur la base de ces études, l’autorisation de mise sur le marché du rituximab pour le traitement initial du pemphigus sévère et modéré a été obtenue en juin 2018 aux États-Unis [2,3]. Une autorisation de mise sur le marché en Europe est attendue dans les prochaines années.
Les premiers résultats d’études sur l’immunoadsorption (IA) (tab. 3) dans le pemphigus ont été publiés en 2007 [4]. Depuis, plus de 100 patients ont été publiés avec différents protocoles thérapeutiques. Toutes les séries de cas ont montré, contrairement au rituximab, une diminution rapide des auto-anticorps et une amélioration clinique rapide, mais aussi un taux élevé de récidive à court terme. L’association de l’AI et du rituximab a permis d’obtenir des rémissions rapides et durables [5].
Des séries de cas et des rapports de cas individuels ont rapporté une bonne réponse après plusieurs séries de traitement par immunoglobulines intraveineuses à haute dose ( IVIG) (tableau 4) en association avec des glucocorticostéroïdes dans le pemphigus vulgaire et le pemphigus paranéoplasique en deuxième ou troisième ligne de traitement après l’échec d’un traitement immunosuppresseur combiné. Ce n’est qu’après plusieurs mois de traitement qu’une réponse a été observée.
Pemphigoïde bulleuse
Le traitement de la pemphigoïde bulleuse doit être adapté à l’activité de la maladie et aux comorbidités de chaque patient. En général, on utilise des méthodes thérapeutiques plus douces que pour les pemphigus. Dans la pemphigoïde bulleuse localisée et modérée, des traitements désinfectants et anti-inflammatoires topiques à base de propionate de clobétasol deux fois par jour sont souvent suffisants. Cependant, le traitement topique de grandes surfaces deux fois par jour est souvent impraticable chez les patients âgés. Un traitement systémique est généralement nécessaire. Le traitement systémique avec initialement 0,5 mg/kg/j d’équivalent prednisolone en combinaison avec des immunosuppresseurs adjuvants tels que l’azathioprine ou le mycophénolate mofétil, afin d’économiser les glucocorticoïdes, constitue le traitement de premier choix en plus du traitement topique (tab. 1). Les autres traitements adjuvants décrits sont le méthotrexate (15 mg/semaine), la dapsone (100 mg/j, tab. 5) et la tétracycline (200 mg/j). L’immunoadsorption (tableau 3) a permis de montrer, dans une série de cas portant sur 20 patients, la diminution rapide des taux d’autoanticorps de la pemphigoïde bulleuse dans le sérum et l’effet thérapeutique qui en découle [6].
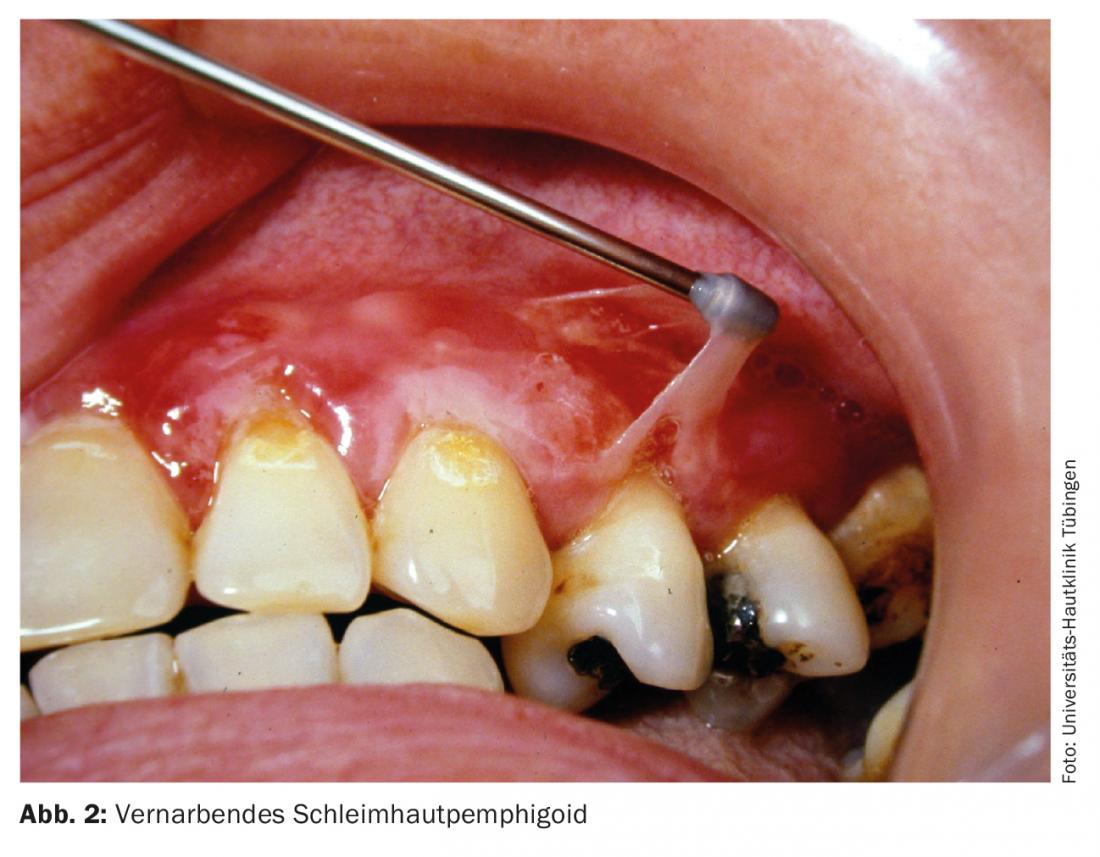
Pemphigoïde muqueuse
Cette dermatose auto-immune est également appelée pemphigoïde cicatricielle. La maladie se caractérise par une grande hétérogénéité clinique, avec ou sans cicatrices. Toutes les muqueuses à épithélium squameux (muqueuse buccale, conjonctives, nasopharynx, œsophage, vulve et rectum) peuvent être touchées. Le traitement dépend de la sévérité clinique, il faut avant tout prévenir les cicatrices (fig. 2). Outre les glucocorticoïdes à haute dose associés à des immunosuppresseurs adjuvants, un traitement par cyclophosphamide (tab. 1), IgIV (tab. 4) ou rituximab (tab. 2) peut également être recommandé chez les patients réfractaires [7].
Dermatose linéaire à IgA
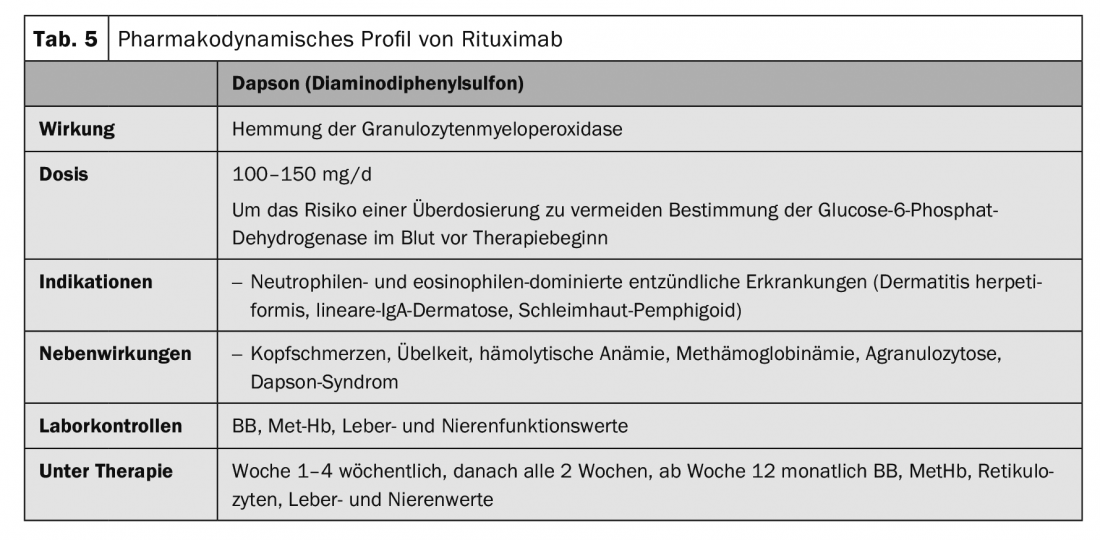
Les agents de première intention dans les dermatoses à IgA linéaires sont les glucocorticostéroïdes et la dapsone (tableau 5). Les glucocorticostéroïdes sont beaucoup moins efficaces dans cette maladie que dans le pemphigus ou la pemphigoïde bulleuse. L ‘immuno-adsorption (tab. 3), les IgIV (tab. 4) et le rituximab (tab. 2) [8,9] ont été décrits comme efficaces dans des rapports de cas.
Epidermolyse bulleuse acquise
L’épidermolyse bulleuse acquise présente dans de nombreux cas une résistance marquée au traitement. La forme mécanobulleuse, en particulier, est souvent résistante au traitement par glucocorticostéroïdes en association avec des immunosuppresseurs adjuvants tels que l’azathioprine ou le mycophénolate-mofétil. (Tab.1). Dans une série de cas portant sur 3 patients et dans des rapports de cas individuels, il a été démontré que Rituximab (Tab. 2) en monothérapie, donne des rémissions de longue durée chez plus de la moitié des patients et en association avec les IgIV (tableau 4) ou l’immunoadsorption (tableau 3) chez plus de 75% des patients [10].
Dermatite herpétiforme de Duhring
En plus de la maladie de peau, une entéropathie sensible au gluten (maladie cœliaque) est toujours présente simultanément, bien que plus de 80% des patients ne présentent pas de symptômes gastro-intestinaux, mais présentent généralement une atrophie villositaire subclinique ou une inflammation du jéjunum. La thérapie est basée sur un régime sans gluten à vie. Un régime sans gluten prévient également l’apparition de lymphomes non hodgkiniens et probablement de cancers du tube digestif. L’effet du régime sans gluten n’apparaît sur la peau qu’après plusieurs mois, de sorte que la dapsone (tableau 5) doit toujours être utilisée initialement. L’entéropathie sous-jacente n’est pas affectée par la dapsone. En cas d’intolérance à la dapsone, la sulfasalazine ou la colchicine peuvent être administrées.
Résumé
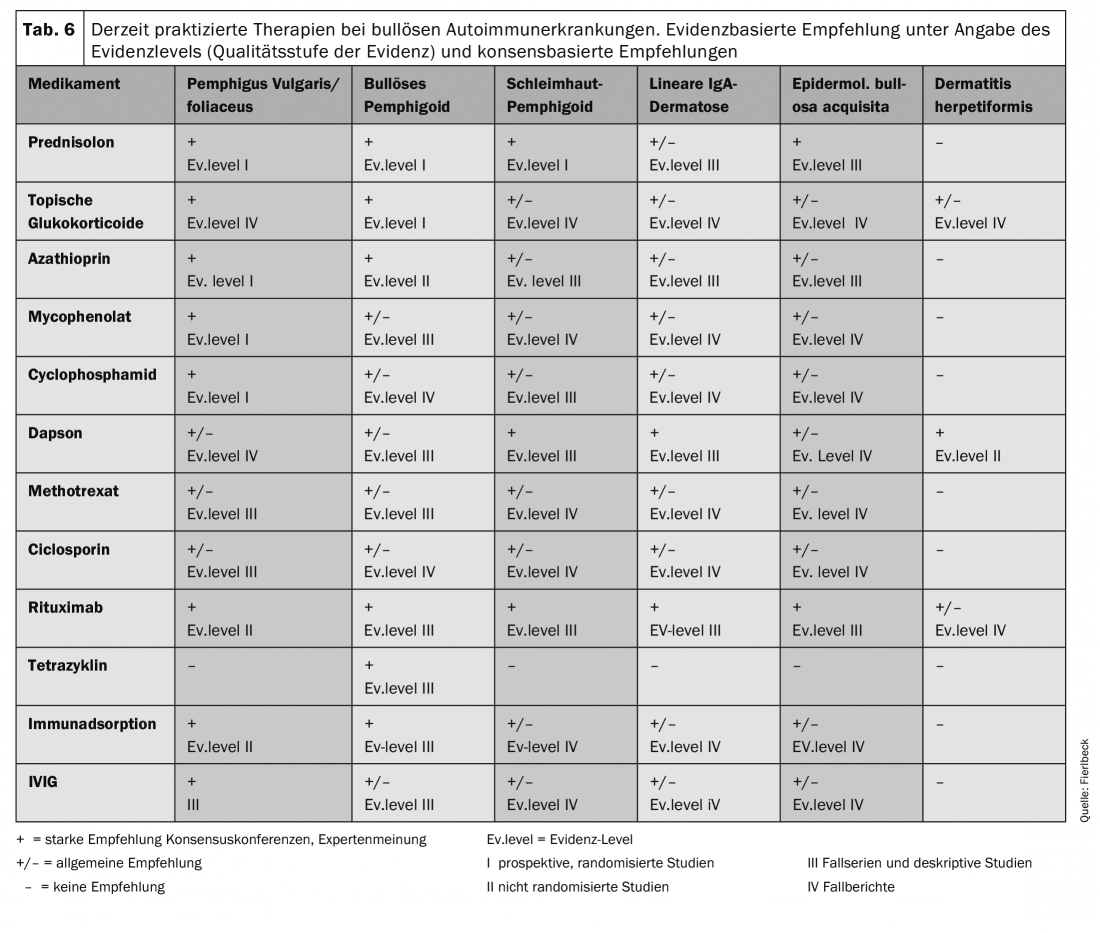
Le tableau 6 donne un aperçu des thérapies actuellement pratiquées . Les recommandations thérapeutiques sont basées sur des études cliniques (niveau de preuve) et des recommandations générales (conférences de consensus, avis d’experts).
Messages Take-Home
- Les glucocorticostéroïdes locaux et oraux associés à l’azathioprine sont des médicaments de première intention, à l’exception de la dermatite herpétiforme.
- En cas de pemphigoïde bulleuse, il est possible de faire un essai avec des glucocorticostéroïdes topiques puissants. Le plus souvent, un traitement oral par de faibles doses de glucocorticostéroïdes en association avec l’azathioprine est nécessaire.
- Le rituximab est indiqué dans les dermatoses bulleuses auto-immunes sévères et réfractaires. En raison de l’effet retardé du rituximab, il convient d’associer initialement des glucocorticostéroïdes.
- L’immuno-adsorption entraîne une amélioration clinique en quelques jours, mais une immunosuppression supplémentaire est nécessaire en raison de la remontée rapide des auto-anticorps et de l’aggravation clinique.
- En cas de dermatite herpétiforme, un régime sans gluten à vie est nécessaire, même en l’absence d’entéropathie manifeste. La dapsone est le traitement de première intention. Les effets du régime sans gluten ne se font sentir qu’au bout de plusieurs mois, alors que la dapsone agit en quelques jours.
Littérature :
- STIKO : communiqué de la Commission permanente sur les vaccinations (STIKO) du RKI. Justification scientifique de la recommandation de vaccination par le vaccin subunitaire contre l’herpès zoster. Institut Robert Koch (RKI), Bulletin épidémiologique 50, 13.12.2018. www.rki.de
- Jolly P, et al : First-line rituximab combined with short-term prednisolon alone for the treatment of pemphigus. Lancet 2017 ; 389(10083) : 2031-2040. dx.doi.org/10.1016/SO140-6736(17)30070-3
- Murrell DF, et al : Diagnostic et prise en charge du pemphigus : recommandation d’un panel international d’experts. J Am Acad Dermatol 2018 Feb 10. pii : S0190-9622(18)30207-X. doi : 10.1016/j.jaad.2018.02.021. [Epub ahead of print]
- Zillikens D, et al. : Recommandation pour l’utilisation de l’immunophérèse dans le traitement de la maladie bulleuse auto-immune. JDDG 2007 ; 5(10) : 366-373.
- Behzad M, et al : Combined treatment with immunoadsorption and rituximab leads to fast and prolonged clinical remission in pemphigus vulgaris. Br J Dermatol 2012 ; 166(4) : 844-852.
- Hübner F, et al : Adjuvant treatment of severe/refractory bullous pemphigoid with protein A immunoadsorption. JDDG 2008 ; 16(9) : 1109-1119.
- Le-Roux-Viller C, et al : Rituximab pour les patients atteints de pemphigoïde à membrane muqueuse réfractaire. Arch Dermatol 2011 ; 147(7) : 843-849.
- Enk A, et al : Utilisation des immunoglobulines à haute dose en dermatologie. JDDG 2009 ; 7(9) : 806-812.
- Pinard C, et al : Dermatose bulleuse linéaire à IgA traitée par rituximab. JAAD Case Rep. 2019 ; 5(2) : 124-126. doi : 10.1016/j.jdcr.2018.11.004
- Bevans SL, Sami N : The Use of Rituximab in treatment of epidermolysis bullosa acquisita : Three new cases and a review of the literature. Dermatol Ther 2018 ; 31(6):e12726. doi : 10.1111/dth12726 [Epub 2018 Oct 3]
DERMATOLOGIE PRATIQUE 2019 ; 29(2) : 21-27









