Le syndrome de Lynch, également connu sous le nom de HNPCC (carcinome colorectal héréditaire non polyposique), est la cause héréditaire la plus fréquente de cancer colorectal dans le monde. Outre les opérations préventives, l’aspirine est également utilisée pour la prévention du cancer. Quand faut-il penser à la présence d’un syndrome de Lynch et que faire concrètement en cas de suspicion ?
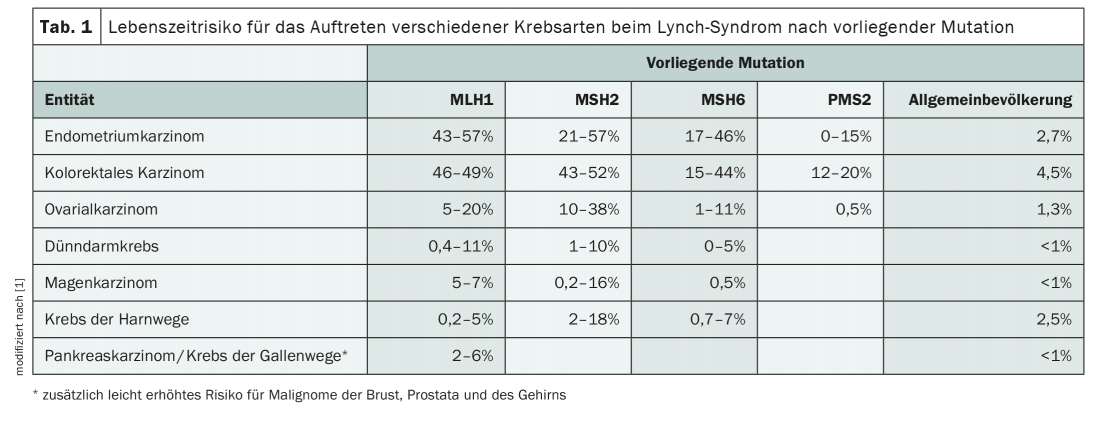
Le syndrome de Lynch, transmis selon un mode autosomique dominant, augmente substantiellement le risque de développer différents types de cancer. Outre les cancers de l’endomètre et les cancers colorectaux, d’autres affections malignes telles que les tumeurs ovariennes, pancréatiques ou gastriques sont également fréquentes (tableau 1). Bien que les mutations à l’origine du syndrome HNPCC soient rares, avec une prévalence de 1:270 à 1:440 dans la population, elles représentent la prédisposition héréditaire au cancer la plus fréquente [1]. Un diagnostic précoce et des mesures préventives impliquant l’ensemble de la famille peuvent prévenir les maladies malignes.
Réparation défectueuse de l’ADN
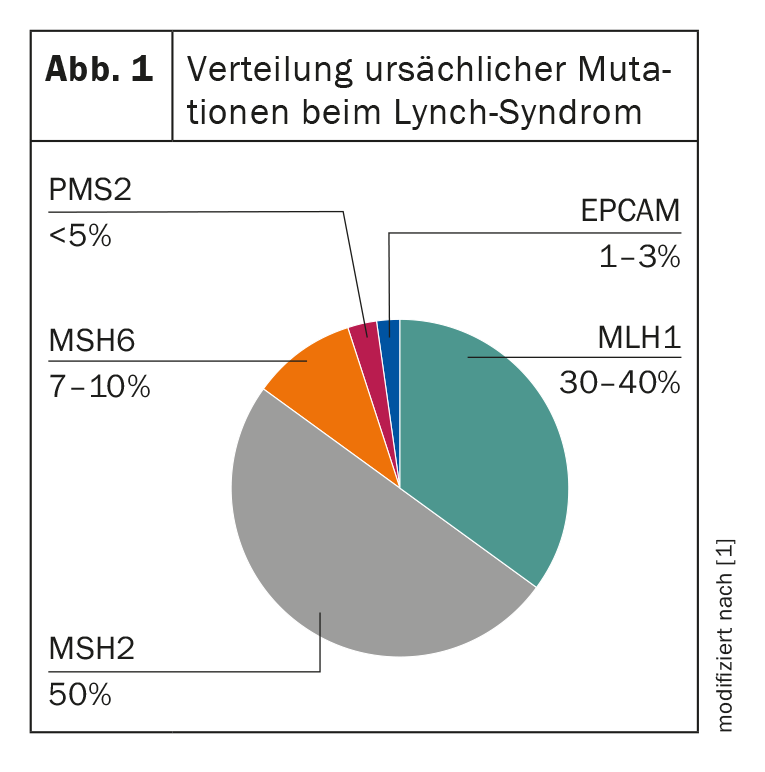
Chez les personnes atteintes, on peut mettre en évidence un défaut génétique de réparation de l’ADN qui se traduit par un allongement des segments répétitifs de l’ADN, appelés microsatellites. Celles-ci diffèrent entre les tissus tumoraux et les tissus sains chez les patients HNPCC, ce que l’on appelle “instabilité des microsatellites” (MSI). La présence d’une telle MSI associée à une suspicion clinique indique donc très probablement un syndrome de Lynch [2]. Jusqu’à présent, on connaît quatre gènes de réparation des mésappariements d’ADN (MMR) dont les mutations de la lignée germinale conduisent au développement du HNPCC : MLH1, MSH2, MSH6 et PMS2. En outre, une délétion de la lignée germinale du gène EPCAM, qui entraîne également une perte de la protéine MSH2 dans la tumeur, peut être considérée comme un facteur causal (Fig. 1). Une perte d’expression correspondante dans les cellules tumorales peut être représentée par immunohistochimie et le diagnostic peut ensuite être confirmé par génétique moléculaire [2]. L’identification de la mutation familiale revêt une grande importance, car elle concerne non seulement le patient lui-même, mais aussi toute sa famille. Elle permet un dépistage prédictif des proches en bonne santé, ce qui rend possible des mesures préventives. Le risque de développer la maladie varie en fonction de la mutation présente et de l’âge. Celui-ci peut être calculé individuellement en utilisant la base de données prospective du syndrome de Lynch [3].
En raison du mode de transmission autosomique dominant, la lignée germinale contient généralement une copie défectueuse du gène responsable, qui a 50 % de chances d’être transmise. Si des mutations somatiques, c’est-à-dire aléatoires, se produisent dans la deuxième copie du gène – qui était fonctionnelle à l’origine -, il en résulte le défaut de réparation de l’ADN décrit ci-dessus et donc une dégénérescence maligne accélérée. Pour qu’un cancer se développe, il faut donc des événements défavorables supplémentaires. Ce mécanisme explique l’augmentation du nombre de cancers, en particulier colorectaux, la dynamique de formation des adénomes étant probablement un facteur de risque indépendant [4].
Le loup déguisé en mouton
Mais quand un test génétique est-il utile ? Quels sont les signes cliniques qui suggèrent la présence d’un syndrome de Lynch ? Contrairement à d’autres syndromes tels que la polypose adénomateuse familiale (PAF), le syndrome HNPCC ne présente pas de caractéristiques phénotypiques claires faisant penser à une maladie héréditaire. Il n’y a généralement que des adénomes ou des carcinomes isolés, qui ne se distinguent pas cliniquement des tumeurs sporadiques. Ce n’est donc qu’avec l’apparition de plusieurs tumeurs malignes, en raison du jeune âge souvent des personnes concernées ou d’une anamnèse familiale frappante, qu’un syndrome de Lynch peut être suspecté [5]. Ainsi, l’âge moyen au moment du diagnostic du cancer colorectal est de 45 ans et dans environ 30% des cas, une autre tumeur typique s’ajoute dans les dix ans [6]. En général, les tumeurs associées au HNPCC sont généralement des adénocarcinomes, et dans l’intestin, elles apparaissent de préférence dans l’hémicolon droit.
Pour faciliter l’identification des patients à risque, plusieurs scores diagnostiques ont été développés au fil des ans, notamment les critères d’Amsterdam et les lignes directrices de Bethesda. Il existe en outre le modèle PREMM pour le calcul des probabilités. L’élément central de tous ces systèmes est une anamnèse familiale détaillée. Pour remplir les critères d’Amsterdam II, qui sont suffisants pour diagnostiquer un HNPCC, au moins trois parents doivent présenter un cancer associé au syndrome de Lynch, au moins deux générations consécutives doivent être touchées et au moins un membre de la famille doit être atteint avant l’âge de 50 ans (aperçu 1) [7]. Dans les cas moins évidents, les lignes directrices de Bethesda aident à identifier les personnes potentiellement atteintes et à les orienter vers un diagnostic approprié. (Aperçu 2). Avec la disponibilité croissante de l’analyse MSI, qui est aujourd’hui déjà réalisée en routine pour de nombreux cancers, ces lignes directrices perdent certes de leur importance, mais elles restent pertinentes dans l’évaluation clinique des résultats [7]. En effet, l’instabilité des microsatellites ne prouve pas le syndrome de Lynch, puisqu’elle est présente dans 10 à 15% des cancers du côlon et dans 15 à 20% des cancers de l’endomètre [5].

Instabilité des microsatellites détectée… et maintenant ?
Si une instabilité des microsatellites ou une déficience des protéines MMR est mise en évidence dans la tumeur d’un patient chez qui un syndrome de Lynch est suspecté, l’indication d’une recherche génétique de mutations déclenchantes est posée. Ces derniers, ainsi que le conseil génétique, sont des prestations obligatoires de la caisse d’assurance maladie, pour autant que les conditions d’une analyse génétique soient remplies [1]. Si une mutation pour laquelle le lien avec la maladie est clairement établi est trouvée, le dépistage ciblé des membres de la famille devrait également être recommandé. Dans ce cadre, les modifications génétiques d’importance (encore) inconnue posent problème. Dans ce cas, la prévention est basée sur l’histoire personnelle de la maladie et les antécédents familiaux, et non sur le résultat de l’analyse génétique. La procédure est donc la même que pour les résultats normaux, les membres de la famille ne doivent pas être testés.
En l’absence de traitements spécifiques, le traitement des tumeurs malignes suit en premier lieu les recommandations qui s’appliquent également aux tumeurs sporadiques. Toutefois, dans les stades avancés, les inhibiteurs de points de contrôle sont de plus en plus utilisés. Par exemple, le pembrolizumab est approuvé en monothérapie pour le traitement de première ligne du cancer colorectal métastatique avec une MSI élevée ou une réparation défectueuse des mésappariements de l’ADN [8]. Des données intéressantes sont attendues dans les années à venir concernant l’utilisation d’autres produits immunothérapeutiques et la valeur de l’instabilité des microsatellites en tant que marqueur prédictif.
Prévention et détection précoce
La détection génétique d’un syndrome de Lynch donne lieu à une longue liste de recommandations pour le dépistage ultérieur du cancer. Elles s’appliquent également aux parents porteurs de la mutation en question. Outre un mode de vie sain et des mesures de dépistage précoce, la chirurgie préventive et les médicaments sont également utilisés. Des coloscopies et des examens gynécologiques réguliers doivent permettre de prévenir les cancers colorectaux et de l’endomètre. Il est également recommandé de procéder à un dépistage de l’Helicobacter pylori (aperçu 3). Il n’existe actuellement aucun dépistage efficace du cancer de l’ovaire et des tumeurs urothéliales, qui sont également fréquents. Il est d’autant plus important de sensibiliser les personnes concernées aux symptômes correspondants et de mettre en œuvre des mesures de surveillance supplémentaires en fonction des tumeurs présentes dans la famille.
Dans certains cas, une hystérectomie prophylactique avec ou sans annexectomie est réalisée après la fin du planning familial. Des résections coliques élargies sont également parfois utilisées. En fin de compte, les avantages de la chirurgie préventive dans le syndrome de Lynch ne sont pas clairs et doivent être évalués individuellement [1]. Il n’existe pas non plus de données solides sur la prise prophylactique d’aspirine, notamment en ce qui concerne la dose et la durée d’administration. Selon les résultats à long terme de l’étude CAPP II, la prise quotidienne de 600 mg d’acide acétylsalicylique après une période de latence d’environ quatre ans réduit significativement le risque de toutes les tumeurs malignes associées au HNPCC. Cet effet semble persister pendant une dizaine d’années après deux ans d’administration, même en l’absence de toute autre administration [9]. Une étude est en cours avec CAPP III pour évaluer des doses plus faibles.
Alors que le diagnostic génétique et la prise en charge des patients atteints du syndrome de Lynch ont beaucoup évolué au cours des dernières années, en particulier dans les centres, le syndrome reste probablement sous-diagnostiqué [10]. Pour contribuer de manière significative à une meilleure prise en charge des personnes concernées, nous pouvons donc avant tout faire une chose : Y penser.
Littérature :
- SAKK : Guide de consultation sur le syndrome de Lynch 2021. www.sakk.ch/sites/default/files/2020-11/Leitfaden%20Lynch-Syndrom.pdf. (dernier accès le 08.05.2021)
- UKB, Institut de génétique humaine : HNPCC / Syndrome de Lynch. www.humangenetics.uni-bonn.de/de/beratung/erbliche-tumorerkrankungen/krankheitsbilder/hnpcc-lynch-syndrom (dernier accès le 08.05.2021)
- Base de données prospective sur le syndrome de Lynch (PLSD) – risque cumulé de cancer en fonction de l’âge, de la variante génétique et du sexe chez les porteurs soumis à une coloscopie. www.plsd.eu.
- Engel C, et al : Efficacité de la surveillance coloscopique annuelle chez les individus atteints de cancer colorectal héréditaire non polyposique. Clin Gastroenterol Hepatol. 2010 ; 8(2) : 174-182.
- Steinke V, et al : Cancer colorectal héréditaire sans polypose. Dtsch Arztebl International. 2013 ; 110(3) : 32-8.
- Lynch HT, et al. : Review of the Lynch syndrome : history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009 ; 76(1) : 1-18.
- Livstone EM : Syndrome de Lynch 2019. Manuel MSD. www.msdmanuals.com/de/profi/gastrointestinale-erkrankungen/tumoren-des-gastrointestinaltrakts/lynch-syndrom (dernier accès le 08.05.2021)
- Information sur les médicaments de swissmedic, Institut suisse des produits thérapeutiques. www.swissmedicinfo.ch (dernier accès le 08.05.2021)
- Burn J, et al. : Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study : a double-blind, randomised, placebo-controlled trial. The Lancet. 2020 ; 395(10240) : 1855-1863.
- Morrow A, et al : Understanding implementation success : protocol for an in-depth, mixed-methods process evaluation of a cluster randomised controlled trial testing methods to improve detection of Lynch syndrome in Australian hospitals. BMJ Open. 2020 ; 10(6) : e033552.
- Vasen HF, et al. : New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroentérologie. 1999 ; 116(6) : 1453-1456.
- Umar A, et al : Lignes directrices révisées de Bethesda pour le cancer colorectal héréditaire non polyposique (syndrome de Lynch) et l’instabilité des microsatellites. J Natl Cancer Inst. 2004 ; 96(4) : 261-268.
InFo ONKOLOGIE & HÉMATOLOGIE 2021 ; 9(3) : 37-39