La tachycardie ventriculaire polymorphe catécholaminergique (CPVT) est une maladie rare des canaux ioniques sans cardiopathie structurelle macroscopiquement détectable. Elle survient exclusivement chez l’enfant et l’adolescent et a une mortalité élevée si elle n’est pas traitée. C’est pourquoi le traitement du cœur structurellement normal est particulièrement important.
La tachycardie ventriculaire polymorphe catécholaminergique (TPVC) est une maladie rare des canaux ioniques sans cardiopathie structurelle macroscopiquement détectable, dont la prévalence est de 1/5000 à 10 000. Décrite pour la première fois en 1975 [1], elle n’a été caractérisée comme une entité distincte qu’en 1995 [2]. Il se caractérise par l’apparition d’extrasystoles ventriculaires polymorphes et de tachycardies ventriculaires (TV) ainsi que de TV bidirectionnelles lors d’un effort physique ou psychologique alors que l’ECG de repos est normal. Bien qu’il existe des rapports de cas isolés de première manifestation symptomatique jusqu’à l’âge de 40 ans, il s’agit principalement d’une maladie des enfants et des adolescents. Selon le sous-type génétique, la première manifestation se produit entre 2 et 20 ans, avec une prévalence familiale dans 30% des cas. Les hommes et les femmes sont touchés à égalité, avec une présentation plus précoce chez les hommes [3]. Un dépistage familial devrait toujours être effectué [4].
Physiopathologie
Bien que de grands progrès aient été réalisés depuis la première description dans la caractérisation des mécanismes sous-jacents de la maladie, ceux-ci ne sont toujours pas totalement élucidés.
Des défauts dans la libération diastolique du calcium (Ca2+) à partir du réticulum sarcoplasmique (RS) entraînent une surcharge en Ca2+ dans la cellule myocardique. Chez les sujets sains, un afflux de Ca2+ extracellulaire via les canaux de type L, déclenché par le potentiel d’action, entraîne une libération de Ca2+ déclenchée par le SR par activation du récepteur de la ryanodine (RYR2). Cela entraîne la contraction myocardique et la phase de plateau du potentiel d’action, suivie d’un retour du Ca2+ dans l’espace extracellulaire et dans le SR. Cependant, dans le cas de la CPVT, un défaut autosomal dominant de RYR2, entre autres, entraîne une libération spontanée et diastolique de Ca2+ à partir du SR. Cela entraîne une inversion du transport de Ca2+ dans la cellule et des post-dépolarisations tardives, qui constituent la base de la CPVT. Si une stimulation β-adrénergique due au stress se produit en plus, les extrasystoles ou tachycardies ventriculaires polymorphes caractéristiques se développent [5].
Outre les mutations du gène RYR2, qui sont détectées chez environ 50 à 55% des patients atteints de CPVT, il existe d’autres mutations de la calsequestrine cardiaque (CASQ2) et de la triadine (TRDN), deux autres composants du budget cellulaire en Ca2+ [5]. En outre, un dysfonctionnement des canaux potassiques (KCNJ2) peut également entraîner un CPVT. Selon le défaut génétique détecté, on peut distinguer plusieurs sous-types. Cependant, dans l’ensemble, les défauts génétiques responsables ne peuvent être détectés que chez environ 60% des personnes atteintes [5].
Diagnostic
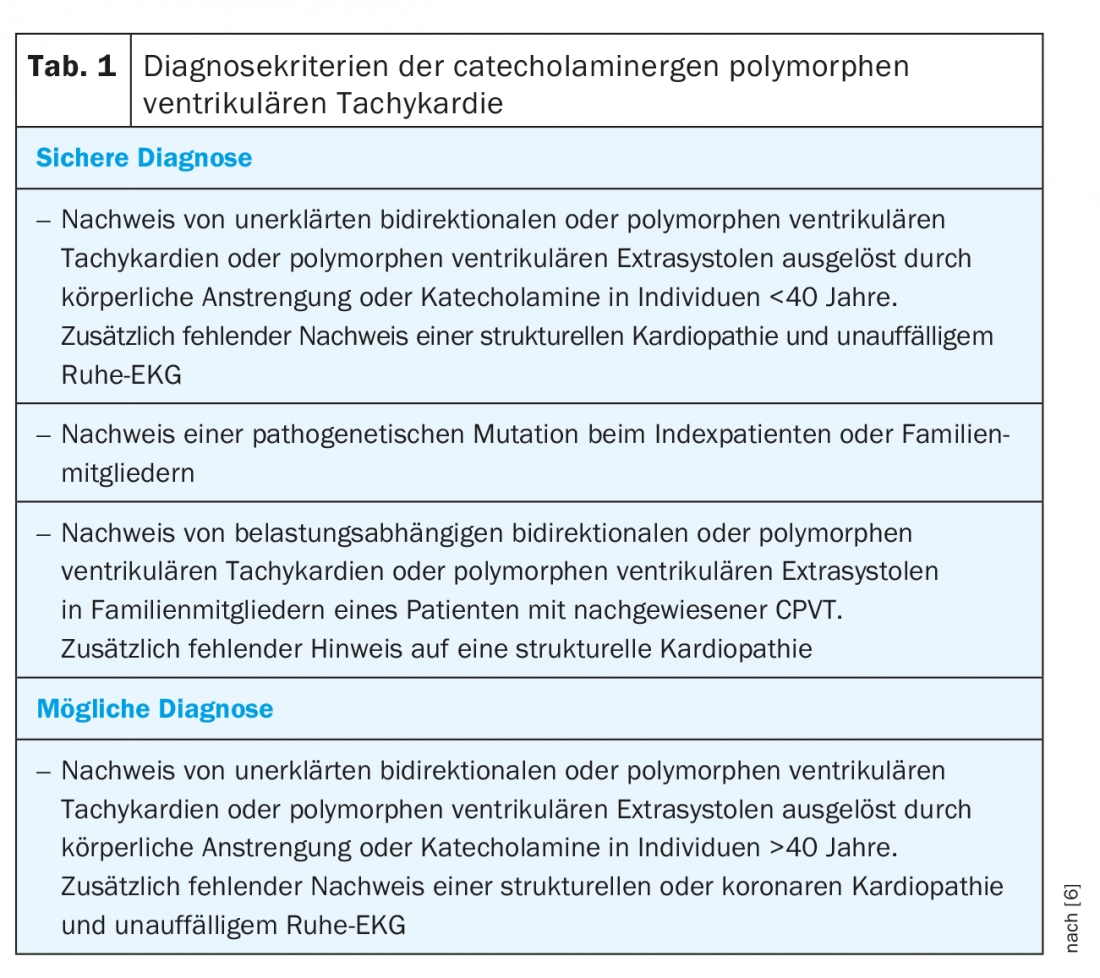
Les critères de diagnostic des sociétés savantes sont résumés dans le tableau 1 [6]. Cependant, il est beaucoup plus difficile d’identifier les personnes susceptibles d’être touchées que d’appliquer les critères de diagnostic. Les présentations cliniques typiques sont des enfants âgés de 2 à 20 ans ayant fait une syncope ou ayant survécu à une mort subite d’origine cardiaque (MSC) pendant un effort physique ou émotionnel [2]. Comme les syncopes dues au CPVT peuvent également entraîner des convulsions et de l’incontinence, les enfants sont souvent traités à tort pour une épilepsie. Le CPVT est alors diagnostiqué tardivement en l’absence d’efficacité des antiépileptiques. De même, une accumulation familiale de syncopes liées à l’effort ou de SCD, ainsi que d’épilepsies familiales résistantes au traitement, doit faire penser à un possible CPVT familial. Une présentation atypique dans le cadre d’un dépistage cardiovasculaire par ergométrie est possible à l’âge adulte, mais beaucoup plus rare.
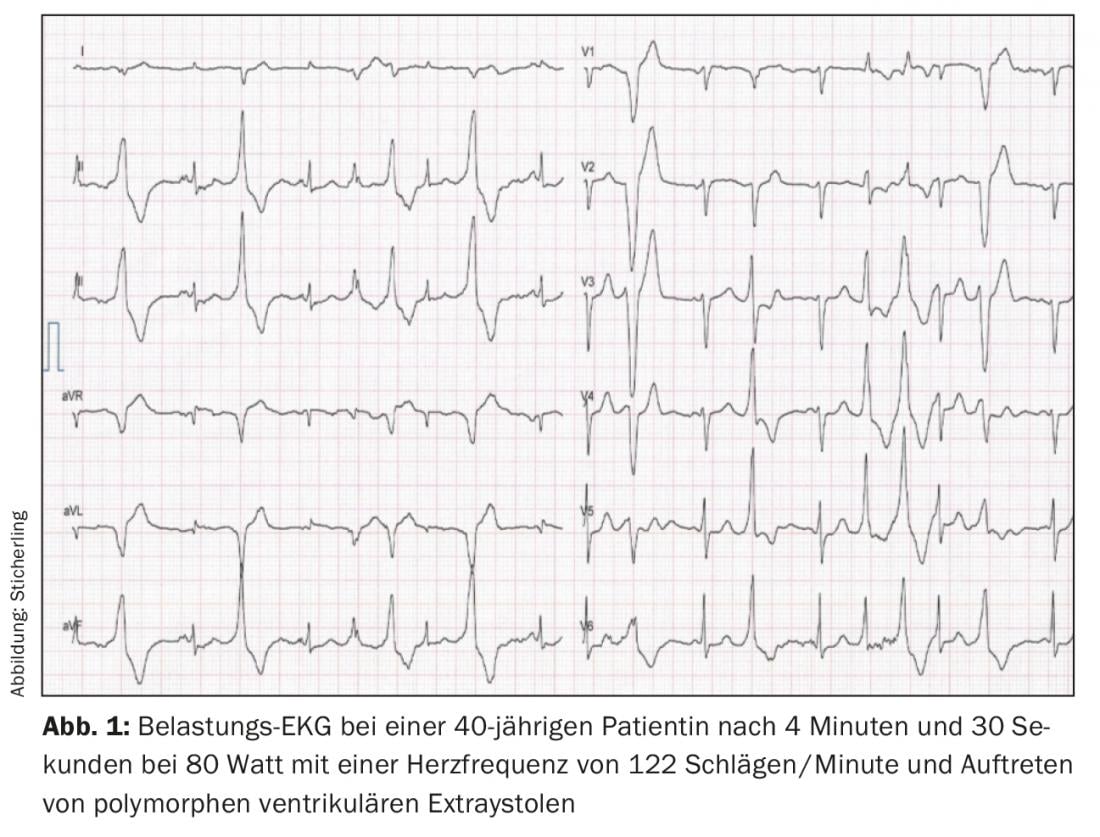
L’ergométrie à l’effort constitue l’étalon-or pour le diagnostic. On constate typiquement un seuil de fréquence cardiaque reproductible individuellement entre 110-130 battements/minute, à partir duquel apparaissent les premières extrasystoles ventriculaires isolées avec un intervalle de couplage d’environ 400 ms. Les extrasystoles présentent principalement un axe supérieur avec un bloc de branche gauche ou un axe inférieur avec un bloc de branche droit [4,7]. Avec l’augmentation de l’effort et de la fréquence cardiaque, les extrasystoles monomorphes sont plus fréquentes, suivies de bigemini et enfin d’extrasystoles polymorphes et/ou de TV non continues (Fig. 1). Dans de rares cas, des TV polymorphes persistantes ou une fibrillation ventriculaire peuvent survenir. L’épreuve d’effort doit donc être arrêtée en cas d’augmentation des symptômes arythmogènes ou d’augmentation de la durée des TV non soutenues [7,8]. Un ECG ambulatoire de longue durée peut avoir une valeur diagnostique supplémentaire chez les patients très jeunes, chez les patients qui ne peuvent pas effectuer d’ECG d’effort ou chez les patients dont l’ECG d’effort est négatif mais dont la suspicion de CPVT persiste. La sensibilité est toutefois inférieure à celle de l’ECG d’effort [9]. Une autre possibilité de diagnostic est la perfusion de catécholamines. Toutefois, sa sensibilité est inférieure à celle de l’épreuve d’effort (environ 75%), de sorte qu’elle ne doit être utilisée à des fins de diagnostic que dans des cas exceptionnels [10]. L’examen électrophysiologique avec stimulation programmée n’a pas de place dans le diagnostic du CPVT [2,10]. Comme indiqué précédemment, l’ECG de repos et l’imagerie médicale n’apportent aucune valeur diagnostique. Des résultats normaux à ces examens sont toutefois obligatoires pour pouvoir poser le diagnostic [6].
Le diagnostic différentiel doit être posé avec d’autres maladies des canaux ioniques comme le syndrome du QT long, en particulier en cas de SCD pendant l’exercice physique comme la natation [11]. Un ECG d’effort peut démasquer un allongement de l’intervalle QTc pendant la phase de récupération, qui n’est pas détectable sur l’ECG de repos, et peut ainsi contribuer à différencier le CPVT [12]. Un autre diagnostic différentiel est le syndrome d’Andersen-Tawil, qui, tout comme un sous-type de CPVT, est associé à une mutation dans le KCNJ2 et peut également se présenter avec des tachycardies ventriculaires bidirectionnelles [13]. Outre les signes phénotypiques de paralysie périodique et de dysmorphie des membres, qui ne sont pas toujours présents, l’ECG d’effort peut ici aussi contribuer à distinguer la CPVT. La différenciation est importante, car les patients atteints du syndrome d’Andersen-Tawil ont un pronostic plus bénin [13]. Outre les maladies des canaux ioniques, il faut toujours penser aux maladies structurelles non encore diagnostiquées telles que les cardiomyopathies arythmogènes, hypertrophiques, ischémiques ou valvulaires, qui sont globalement beaucoup plus fréquentes. Les autres causes de TV bidirectionnelle sont une intoxication à la digoxine ou une myocardite [14,15].
Thérapie
Le traitement de la CPVT consiste en des modifications du mode de vie, un traitement médicamenteux, une stratification du risque pour un DAI, voire une dénervation sympathique cardiaque du côté gauche (DCSG). Étant donné que la mortalité peut atteindre 50% chez les patients gravement atteints, un traitement adéquat revêt une grande importance [2,16] (tableau 2). En outre, un dépistage familial devrait toujours être effectué en raison de la transmission autosomique dominante du défaut RYR2.
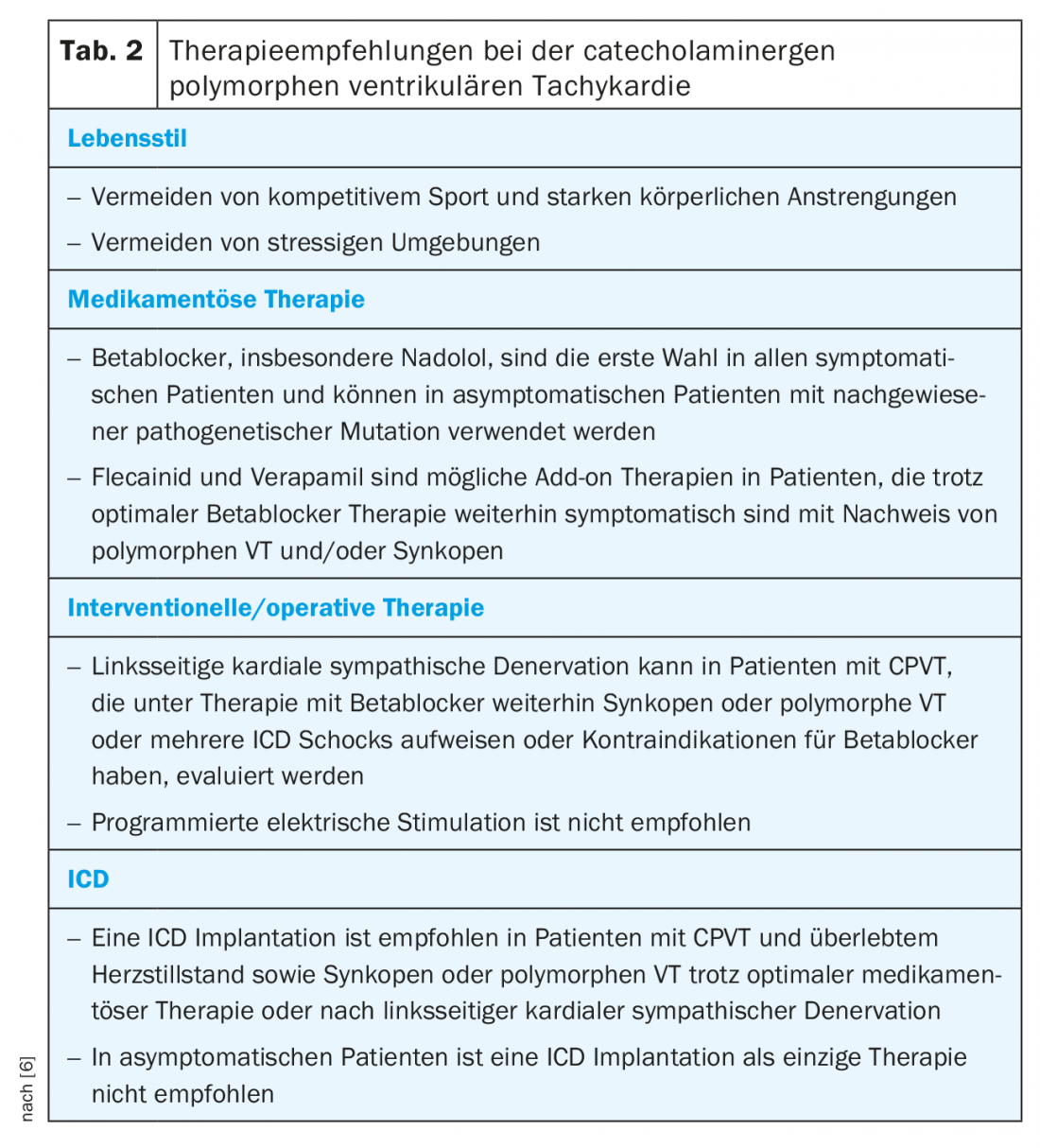
Comme l’effort physique peut déclencher des arythmies ventriculaires, les patients doivent éviter les sports compétitifs. Une petite étude a certes pu montrer que chez les patients atteints de CPVT traités de manière adéquate, le sport compétitif ne provoquait qu’une augmentation des arythmies sans augmentation de la mortalité [17], mais il n’est pas possible de l’appliquer à tous les patients. Un surveillant doit être présent, en particulier pour la natation.
Les bêtabloquants sont la pierre angulaire du traitement médicamenteux, car ils empêchent notamment d’atteindre le seuil de fréquence cardiaque arythmogène. La plupart des données sont disponibles pour le nadolol non sélectif à longue durée d’action. Cependant, ce produit n’est plus disponible dans de nombreux pays, comme la Suisse [16,18,19]. Le carvédilol, qui a un effet démontré sur le RYR2, est une alternative possible [20]. Cependant, les données cliniques chez les patients CPVT font défaut. En principe, il faut toujours procéder à un dosage complet jusqu’à la dose maximale tolérable. Il est important de noter que chez les enfants, la dose doit être adaptée au poids corporel. Un traitement bêtabloquant adéquat permet de réduire le taux de SCD fatal à 6,4% sur 8 ans [19]. De même, les patients génotypiquement positifs, détectés lors d’un dépistage familial mais ne présentant pas d’arythmie à l’ECG d’effort, doivent recevoir un traitement bêtabloquant [6]. Si des arythmies ou des couples et des extrasystoles ventriculaires fréquentes continuent d’être observés à l’ergométrie d’effort sous un traitement adéquat par bêtabloquant, un traitement supplémentaire par flécaïnide doit être évalué. La flécaïnide a également un effet inhibiteur direct sur RYR2 et, dans les essais cliniques, elle a supprimé les arythmies ventriculaires chez 76% des patients déjà traités par bêtabloquants [21,22]. Les bloqueurs des canaux calciques semblent apporter un bénéfice supplémentaire en tant que traitement alternatif complémentaire. De petites études non randomisées ont montré que le vérapamil, en plus des bêtabloquants, réduisait encore la survenue d’arythmies ventriculaires [23,24]. Il n’existe pas encore d’études randomisées de grande envergure sur le vérapamil. Parmi les options thérapeutiques non encore utilisées en clinique, mais potentiellement prometteuses, on trouve la propafénone, qui s’est révélée prometteuse sur un nombre limité de patients [25] et le dantrolène, qui a montré son efficacité dans un modèle cellulaire [26].
Si le traitement médicamenteux ne permet pas de contrôler suffisamment les arythmies, il est possible d’évaluer une LKSD, qui consiste à enlever par thoracoscopie la moitié inférieure du ganglion stellaire gauche et les ganglions thoraciques T2-4. Il en résulte une diminution significative de la sécrétion de noradrénaline dans le cœur, avec de très bons résultats dans de petites séries de patients atteints de CPVT sévère. La complication la plus fréquente de la DLFT est un syndrome de Horner, le plus souvent transitoire, mais qui peut aussi persister dans 2 à 3% des cas. Des complications plus rares sont des lésions des structures environnantes telles que la plèvre, le nerf phrénique, le plexus brachial et les nerfs somatiques, entraînant des douleurs lancinantes dans la région de l’épaule gauche. Comme effet secondaire, il peut y avoir une absence de transpiration de la main gauche et du front gauche avec une peau plus chaude et plus sèche en comparaison avec les côtés [27,28].
L’implantation d’un DAI ne doit être réalisée que chez des patients sélectionnés qui ont survécu à un SCD ou qui continuent à souffrir de syncopes ou de TV polymorphes ou bidirectionnelles malgré un traitement optimal. Si un LKSD est disponible, il doit également être effectué avant l’implantation [6]. Cette recommandation restrictive existe surtout parce que les chocs DAI entraînent en soi une sécrétion supplémentaire de catécholamines. Ceux-ci peuvent alors déclencher d’autres arythmies et entraîner un cercle vicieux, de sorte que le DAI doit être programmé avec des cut-offs élevés et de longs délais avant la délivrance du choc [6,29].
Résumé
Le CPVT est une maladie extrêmement rare qui survient presque exclusivement chez les enfants et les adolescents et qui, en l’absence de traitement, présente un taux de mortalité élevé. Un traitement par bêtabloquant est, avec une adaptation du mode de vie, l’intervention la plus importante en termes de pronostic. L’implantation d’un DAI est associée à des effets proarythmiques particuliers, potentiellement liés à la délivrance d’un choc, et l’indication en prophylaxie primaire doit donc être posée avec réserve. D’autres études sont nécessaires pour mieux comprendre la maladie et trouver de nouvelles approches thérapeutiques.
Messages Take-Home
- La CPVT survient presque exclusivement chez l’enfant et l’adolescent dans des cœurs structurellement normaux et a une mortalité élevée en l’absence de traitement.
- Les principaux traitements sont l’adaptation du mode de vie et les bêtabloquants.
- L’implantation d’un DAI ne doit être effectuée que chez les patients ayant survécu à une mort subite, présentant des syncopes ou des tachycardies ventriculaires polymorphes persistantes malgré un traitement médicamenteux maximal ou après évaluation d’une dénervation sympathique cardiaque gauche.
- La délivrance d’un choc par un DAI présente un risque élevé de proarythmie, de sorte que des temps de détection longs et des fréquences d’intervention élevées doivent être programmés pour le DAI.
Littérature :
- Reid DS, et al : Tachycardie bidirectionnelle chez un enfant. Une étude utilisant His bundle electrography. Br Heart J. 1975 ; 37:339-344.
- Leenhardt A, et al. : Tachycardie ventriculaire polymorphe catécholaminergique chez l’enfant. Un suivi de 7 ans chez 21 patients. Circulation. 1995 ; 91 : 1512-1519.
- Priori SG, et al : Caractérisation clinique et moléculaire des patients atteints de tachycardie ventriculaire polymorphe catécholaminergique. Circulation. 2002 ; 106 : 69-74.
- Leenhardt Antoine, Denjoy Isabelle, Guicheney Pascale. Tachycardie ventriculaire polymorphe catécholaminergique. Circ Arrhythm Electrophysiol. 2012 ; 5 : 1044-1052.
- Pérez-Riera AR, et al : Catecholaminergic polymorphic ventricular tachycardia, an update. Ann Noninvasive Electrocardiol Off J Int Soc Holter Noninvasive Electrocardiol Inc. 2018;23 : e12512.
- HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. 2013 ; 69.
- van der Werf C, Wilde AAM : Catecholaminergic polymorphic ventricular tachycardia : from bench to bedside. Heart. 2013 ; 99 : 497-504.
- Svendsen JH, Geelen P, EHRA Scientific Initiative Committee : Screening for, and management of, possible arhythmogenic syndromes (channelopathies/ion channel diseases). Eur Eur Pacing Arrhythm Card Electrophysiol J Groupes de travail Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2010 ; 12 : 741-742.
- Sy RW, et al : Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Rythme cardiaque. 2011 ; 8:864-871.
- Sumitomo N, et al : Catecholaminergic polymorphic ventricular tachycardia : electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Cœur. 2003;89 : 66-70.
- Tester DJ, et al : Unexplained drownings and the cardiac channelopathies : a molecular autopsy series. Mayo Clin Proc. 2011 ; 86 : 941-947.
- Sy RW, et al. : Dérivation et validation d’un algorithme simple basé sur l’exercice pour la prédiction du test génétique chez les parents de probands LQTS. Circulation. 2011 ; 124 : 2187-2194.
- Inoue YY, et al : Différentes réponses à l’exercice entre le syndrome d’Andersen-Tawil et la tachycardie ventriculaire polymorphe catécholaminergique. Eur Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2018 ; 20 : 1675-1682.
- Berte B, et al : Tachycardie ventriculaire bidirectionnelle dans la myocardite fulminante. Eur Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2008 ; 10 : 767-768.
- Valent S, Kelly P : Images en médecine clinique. Tachycardie ventriculaire bidirectionnelle induite par la digoxine. N Engl J Med. 1997 ; 336 : 550.
- Hayashi M, et al : Incidence et facteurs de risque d’événements arythmiques dans la tachycardie ventriculaire polymorphe catécholaminergique. Circulation. 2009 ; 119 : 2426-2434.
- Ostby SA, et al : Competitive Sports Participation in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia : A Single Center’s Early Experience. JACC Clin Electrophysiol. 2016 ; 2 : 253-262.
- Leren IS, et al : Le nadolol diminue l’incidence et la sévérité des arythmies ventriculaires lors d’un test de stress à l’exercice par rapport aux β-bloqueurs β-sélectifs chez les patients atteints de tachycardie ventriculaire polymorphe catécholaminergique. Rythme cardiaque. 2016 ; 13 : 433-440.
- van der Werf C, Zwinderman AH, Wilde AAM : Approche thérapeutique pour les patients atteints de tachycardie ventriculaire polymorphe catécholaminergique : état de l’art et développements futurs. Eur Eur Pacing Arrhythm Card Electrophysiol J Groupes de travail Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2012 ; 14:175-183.
- Zhou Q, et al : Carvedilol and its new analogs suppress arhythmogenic store overload-induced Ca2+ release. Nat Med. 2011 ; 17 : 1003-1009.
- van der Werf C, et al : Flecainide therapy reduces exercise-induced ventricular arhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011 ; 57 : 2244-2254.
- Watanabe H, et al : Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009 ; 15 : 380-383.
- Rosso R, et al : Bloqueurs de canaux calciques et bêta-bloquants versus bêta-bloquants seuls pour prévenir les arythmies induites par l’exercice dans la tachycardie ventriculaire polymorphe catécholaminergique. Rythme cardiaque. 2007 ; 4 : 1149-1154.
- Swan H, et al : L’antagonisme des canaux calciques réduit les arythmies ventriculaires induites par l’exercice chez les patients atteints de tachycardie ventriculaire polymorphe catécholaminergique avec mutations RyR2. J Cardiovasc Electrophysiol. 2005 ; 16 : 162-166.
- Hwang HS, et al. : Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011 ; 4 : 128-135.
- Jung CB, et al : Dantrolene rescues arhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012 ; 4 : 180-191.
- Odero A, et al. : Dénervation sympathique cardiaque gauche pour la prévention des arythmies fatales : l’approche supraclaviculaire chirurgicale de la sympathectomie cervicothoracique. Rythme cardiaque. 2010 ; 7 : 1161-1165.
- Wilde AAM, et al : Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008 ; 358 : 2024-2029.
- Mohamed U, et al : Mort cardiaque subite malgré un défibrillateur cardioverteur implantable chez une jeune femme atteinte de tachycardie ventriculaire catécholaminergique. Rythme cardiaque. 2006 ; 3 : 1486-1489.
CARDIOVASC 2019 ; 18(2) : 12-15