
L’hypertension pulmonaire est divisée en 5 groupes différents, le groupe 1 reflétant l’hypertension artérielle pulmonaire (HTAP). Les traitements médicamenteux spécifiques à l’HTP autorisés à ce jour interviennent au niveau de trois voies de signalisation différentes (voie du NO, voie de la prostacycline et voie du récepteur de l’endothéline). Depuis l’approbation du premier médicament spécifique à l’HTAP (bosentan) en Europe il y a environ 20 ans, beaucoup de choses ont changé. De nombreuses autres substances ont été ajoutées et ont permis d’améliorer le pronostic des patients atteints d’HTAP.
L’hypertension pulmonaire est divisée en 5 groupes différents, le groupe 1 reflétant l’hypertension artérielle pulmonaire (HTAP). Les traitements médicamenteux spécifiques à l’HTP autorisés à ce jour interviennent au niveau de trois voies de signalisation différentes (voie du NO, voie de la prostacycline et voie du récepteur de l’endothéline). Depuis l’approbation du premier médicament spécifique à l’HTAP (bosentan) en Europe il y a environ 20 ans, beaucoup de choses ont changé. De nombreuses autres substances ont été ajoutées et ont permis d’améliorer le pronostic des patients atteints d’HTAP.
Alors que l’HTAP idiopathique (HTAPI) a longtemps été diagnostiquée en premier lieu chez des patients jeunes et sans comorbidités cliniquement significatives, un changement démographique est apparu ces dernières années. Actuellement, de plus en plus de patients âgés présentant des comorbidités pulmonaires et/ou cardiaques sont diagnostiqués avec une hypertension pulmonaire précapillaire sévère et sont considérés comme des patients IPAH sur la base de la classification actuelle. Il est ressorti de différentes analyses de bases de données [1,2] que les patients IPAH présentent des caractéristiques de maladie très différentes et qu’il existe différents clusters ou phénotypes. Il convient de souligner que la capacité de diffusion (DLCO) est, entre autres, un facteur discriminant et pronostique décisif [3–5].
Cet article reprend la présentation “HTAP : qu’y a-t-il au-delà de la nouvelle ligne directrice”, faite le 30 mars 2023 lors du 63e Congrès allemand des pneumologues à Düsseldorf. La présentation a démontré les données d’étude de patients diagnostiqués avec une IPAH qui présentaient une légère altération de la structure pulmonaire au scanner thoracique et/ou un trouble de diffusion alvéolocapillaire sévère d’étiologie indéterminée.
Les comorbidités comme facteur pronostique – état actuel des études
Déjà en 2016, dans le cadre de la conférence de consensus de Cologne, les experts germanophones de l’HTP ont discuté du fait que la population de patients IPAH inclus dans des études thérapeutiques prospectives, multicentriques et contrôlées par placebo n’est souvent pas comparable aux patients inclus dans différents registres nationaux de l’HTP, qui dominent également dans la pratique clinique quotidienne. Il était déjà évident à l’époque que ces derniers étaient plus âgés, présentaient plus de comorbidités et ressemblaient phénotypiquement aux patients atteints de cardiopathie gauche et/ou de pneumopathie en raison de leurs facteurs de risque, mais qu’ils étaient classés comme IPAH selon les critères de définition également en vigueur actuellement. Les termes de patients IPAH “typiques” et “atypiques” ont été introduits à l’époque pour tenir compte de cette évolution. L'”IPAH typique” était alors définie sur le plan hémodynamique, après exclusion de toutes les causes connues d’HTP, par une pression artérielle pulmonaire moyenne (PAPm) ≥25 mmHg et une pression artérielle pulmonaire occlusive (PAPO) ≤15 mmHg. En outre, comme dans l’étude AMBITION (ambrisentan plus tadalafil versus ambrisentan ou tadalafil en monothérapie), seuls deux facteurs de risque maximum de dysfonctionnement diastolique du ventricule gauche (Heart Failure with preserved left-ventricular ejection fraction, HFpEF : hypertension artérielle, maladie coronarienne, diabète sucré et obésité avec BMI >30 kg/m2) et la capacité de diffusion (DLCO) doit être d’au moins 45% de la valeur cible. [6,7]. Dans le groupe de l'”IPAH atypique”, dont le profil hémodynamique ne différait pas de celui de l'”IPAH classique”, les patients étaient principalement âgés (généralement >65 ans) et présentaient le profil de risque ou les maladies concomitantes des patients atteints de cardiopathie gauche ou de pneumopathie. Ils étaient alors appelés “patients IPAH de phénotype cardiaque” ou “patients IPAH de phénotype pulmonaire”.
Les patients présentant un phénotype pulmonaire étaient caractérisés par une bodyplethysmographie et un scanner thoracique (presque) normaux, une hypoxémie souvent sévère et une réduction significative de la capacité de diffusion (DLCO <45% de la valeur théorique) (tableau 1). En ce qui concerne le traitement ciblé de l’HTP, une monothérapie était initialement recommandée chez les patients “atypiques”, car l’efficacité et la tolérance des médicaments spécifiques à l’HTP dans ce collectif n’avaient pas été suffisamment étudiées jusqu’à présent pour permettre de formuler des recommandations fondées sur des preuves. Les données du registre ont montré que ces patients étaient principalement traités avec des inhibiteurs de la PDE5 et qu’ils étaient peu enclins à suivre un traitement combiné.

DLCO comme facteur de risque pronostique – état actuel des études
Une étude publiée en 2010 [4] décrivait déjà un lien entre la DLCO et la survie chez les patients atteints d’HTAP. Trois plages de DLCO (DLCO >64%, DLCO 43-63%, DLCO <43%) ont ici été mises en évidence, qui discriminaient bien le risque de décès. Dans cette étude, la diminution de la DLCO était associée de manière multivariée à l’âge des patients, à la présence d’une collagénose, à une capacité fonctionnelle réduite, à la demande en oxygène et à des volumes pulmonaires réduits et des modifications morphologiques au scanner. Ces corrélations étaient indépendantes de l’hémodynamique cardio-pulmonaire. Les patients qui présentaient la plage de DLCO la plus basse (<43%) avaient un risque de décès 2,7 fois plus élevé.
En 2013, Trip et al. [8] que les patients IPAH avec une DLCO <45% ont un pronostic significativement plus mauvais par rapport aux patients IPAH avec une DLCO corrigée en fonction de l’âge plus élevée. Au total, environ 75% des patients atteints d’IPAH présentaient une DLCO réduite (principalement légère à modérée). L’analyse excluait les patients présentant des causes connues de réduction sévère de la diffusion, telles que les foramens ovales ouverts, les collagénoses, les maladies veino-occlusives pulmonaires ( PVOD ), les cardiopathies gauches sévères et les maladies pulmonaires. Dans cette étude, des modifications légères et modérées du scanner ne constituaient pas une exclusion pour le diagnostic d’IPAH.
Trip et al. ont démontré que les patients atteints d’IPAH + DLCO <étaient en moyenne 45% plus âgés (67 ans contre 49 ans), majoritairement de sexe masculin, plus souvent atteints d’une maladie coronarienne et plus souvent fumeurs. Les paramètres fonctionnels respiratoires (VEMS, VEMS/CVF et CLT) avaient tendance à être plus faibles et les modifications morphologiques par scanner (emphysème léger à modéré ou fibrose) étaient plus fréquentes par rapport aux patients dont la DLCO était ≥45%. En revanche, l’hémodynamique pulmonaire était comparable. Néanmoins, ces patients présentaient une moins bonne capacité fonctionnelle à l’effort (test de marche de 6 minutes).
La survie à 3 ans (DLCO <45% vs. DLCO ≥45%) était de 54% et 86% respectivement, et de 30% et 80% à 5 ans. Les auteurs ont conclu de leurs résultats que la réduction sévère de la DLCO était probablement due à un sous-type de PH à considérer séparément, qui pourrait être déclenché par l’inhalation de fumée de cigarette.
En 2017, l’équipe de Hanovre dirigée par Olsson et al. [5] que les patients atteints d’IPAH avec une fonction pulmonaire complètement normale et un scanner thoracique normal, mais avec une DLCO nettement réduite (<45% de la valeur théorique) sont comparables aux patients atteints de CPFE (Combined pulmonary fibrosis and emphysema) en termes de pronostic. Les deux groupes étaient composés principalement d’hommes ayant des antécédents de tabagisme, dont les caractéristiques ne différaient pas, à l’exception des modifications fonctionnelles pulmonaires et morphologiques au scanner.
Tous les patients de cette étude ont été traités avec des inhibiteurs de la PDE5, mais tant la distance de marche au test de marche de 6 minutes que la pression partielle d’oxygène (paO2) ont eu tendance à se détériorer de manière comparable dans les deux groupes sous traitement. La mortalité des deux groupes n’était pas significativement différente à 1, 2 et 4 ans (80% / 76% / 38% vs 64% / 42% / 42%). Les auteurs ont conclu que la cause de la réduction sévère de la DLCO chez leurs patients atteints d’IPAH était probablement une “vaculopathie pulmonaire liée au tabagisme”.
Comme le montrent les données du registre de l’HTP de Sheffield (ASPIRE ; Royaume-Uni), les patients atteints d’HTAP et présentant des modifications parenchymateuses légères au scanner thoracique ne peuvent pas bénéficier d’une amélioration de leur capacité d’exercice malgré un traitement combiné spécifique à l’HTAP. Ces patients présentaient également un pronostic significativement plus mauvais que les patients IPAH sans anomalies tomodensitométriques thoraciques. Chez les patients IPAH sans modification du scanner, mais avec une DLCO <45%, le pronostic ajusté à l’âge dans cette étude de registre était également significativement plus mauvais que chez les patients IPAH avec une DLCO ≥45% [1].
En 2020, les patients IPAH de la base de données européenne COMPERA (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) ont été soumis à une analyse en grappes basée sur les variables suivantes : âge, sexe (masculin vs féminin), statut tabagique ([Ex]Fumeurs : oui vs non), DLCO (<45% vs. ≥45% de la valeur théorique) et la présence vs. l’absence d’au moins un facteur de risque de dysfonctionnement diastolique du ventricule gauche (obésité, hypertension artérielle, maladie coronarienne et diabète sucré). Les clusters trouvés ont été analysés, entre autres, en fonction de leurs caractéristiques de base, de leur réponse aux traitements spécifiques de l’HTP (modifications de la distance de marche de 6 minutes, de la classe fonctionnelle et des taux sanguins de biomarqueurs) et de leur pronostic. Trois clusters différents ont été identifiés (figure 1 ; clusters 1-3).
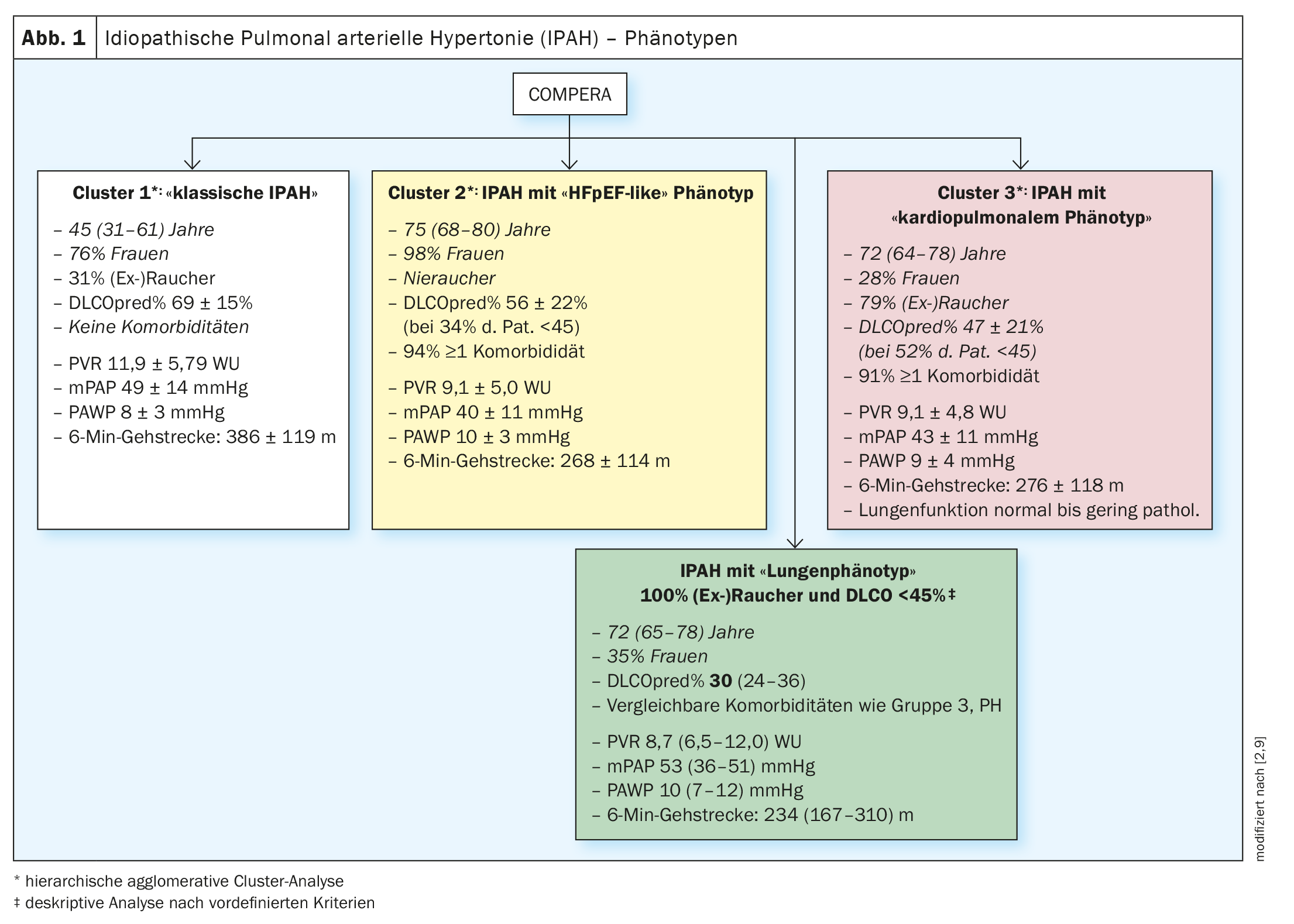
Les trois clusters étaient caractérisés par la présence d’une HTP précapillaire sévère (la PAWP était en moyenne inférieure à 11 mmHg dans les 3 groupes) et une fonction pulmonaire (en grande partie) normale, les patients du cluster 3 présentant comparativement les valeurs les plus basses et l’hypoxémie la plus prononcée pour les paramétriques de la fonction pulmonaire. Après ajustement pour l’âge, les patients du cluster 3 (patients IPAH avec phénotype cardiopulmonaire) avaient un pronostic significativement plus mauvais par rapport aux patients du cluster 1 (patients IPAH sans comorbidités) [2].
Alors que dans cette analyse en cluster, tous les patients du cluster 3 du registre COMPERA n’avaient pas une DLCO <45% (53% des cas) et un statut (ex)tabagique positif (79% des cas), dans une autre étude de registre de l’HTP, une DLCO <45% et un statut (ex)tabagique positif étaient par définition nécessaires pour le diagnostic d’une IPAH avec phénotype pulmonaire [9]. Pour cette étude, la base de données du registre COMPERA a de nouveau été analysée en premier lieu et le registre ASPIRE a servi à valider les résultats de manière indépendante [9].
Cette étude a montré que le groupe de patients IPAH avec un “phénotype pulmonaire” ressemblait beaucoup plus aux patients du groupe 3-PH (HTP associée à une maladie pulmonaire significative) en termes de réponse au traitement et de pronostic qu’aux patients avec une IPAH dite “classique” (IPAH sans facteurs de risque de HFpEF) [9].
L’analyse des données COMPERA a révélé, comme dans l’analyse précédente [2], que les patients atteints d’IPAH “classique” (sans facteurs de risque d’HFpEF) étaient principalement plus jeunes (45 ans en médiane) et généralement des femmes (77%). Environ 1/3 d’entre eux avaient des antécédents de tabagisme avec une médiane de 14 pack years (py), mais une fonction pulmonaire préservée, et une DLCO seulement légèrement réduite (69% en médiane). Sur le plan hémodynamique, ces patients étaient caractérisés par une hypertension pulmonaire précapillaire sévère (mPAP 48 mmHg, PVR 10,9 wood units (WU)) et une capacité fonctionnelle moyennement réduite (distance de marche de 6 minutes : 410 m) [9].
En comparaison, les patients IPAH présentant un phénotype pulmonaire étaient plus âgés (72 ans en médiane) et plus souvent de sexe masculin (65%). Tous les patients présentant un phénotype pulmonaire étaient par définition fumeurs et avaient une médiane de 40 py. La CVF et le VEMS étaient en médiane dans les limites inférieures de la norme ou légèrement réduits et significativement plus bas que chez les patients atteints d’IPAH classique. La DLCO était nettement limitée à 30% en médiane et la paO2était de 56 mmHg en médiane. Par rapport aux patients IPAH classiques, l’hémodynamique était moins compromise (mPAP 43 mmHg, PVR 8,7 WU), mais la capacité fonctionnelle à l’effort (distance de marche de 6 minutes) était significativement plus limitée (234 m). (Fig. 1 – encadré vert).
Dans cette étude, les patients du groupe 3-PH avaient un âge médian comparable à celui des patients présentant un phénotype pulmonaire (71 ans vs. 72 ans en médiane) et une répartition par sexe comparable (63% vs. 65% d’hommes) [9]. 81% étaient des (ex)fumeurs avec une médiane de 40 py, alors qu’il y avait des limitations fonctionnelles respiratoires du VEMS et de la CVF et que la DLCO était la plus réduite avec une médiane de 26%. En revanche, l’hémodynamique était comparativement moins limitée (mPAP 39 mmHg et PVR 7,4 WU en médiane) et la capacité fonctionnelle à l’effort (distance de marche de 6 minutes) était comparable à celle des patients présentant un phénotype pulmonaire (238 m contre 234 en médiane).
Les modifications morphologiques par TDM dans le sens d’un emphysème ou d’une maladie pulmonaire interstitielle dans le groupe de patients ASPIRE ont augmenté de manière significative du groupe des patients IPAH “classiques” vers les patients du groupe 3-PH (de 10% à environ 60%) [9].
Les trois groupes de PH présentaient des différences significatives en termes de réponse au traitement et de pronostic. Chez les patients IPAH classiques, par rapport aux patients IPAH de phénotype pulmonaire et par rapport aux patients IPAH du groupe 3, les probabilités de survie à 3 ans étaient de 90% vs 49% vs 43% et les probabilités de survie à 5 ans étaient de 84% vs 31% vs 26%. Les auteurs ont émis l’hypothèse que la cause de l’insuffisance cardiaque droite chez les patients caractérisés comme ayant une IPAH avec un phénotype pulmonaire était une hypertension pulmonaire causée par la fumée de cigarette et ont conclu que ces patients devraient probablement être classés comme des patients du groupe 3 de l’HTP et que la place des médicaments spécifiques à l’HTP chez ces patients n’était pas claire [9].
Résumé
En résumé, les patients (ex)fumeurs qui répondent aux critères diagnostiques actuels de l’IPAH, mais qui présentent une très forte réduction de la DLCO inexplicable par ailleurs et/ou des anomalies parenchymateuses même minimes au scanner thoracique, pourraient appartenir à un groupe d’HTP différent (le plus probable étant le groupe d’HTP 3) de celui des patients présentant une IPAH classique (groupe d’HTP 1).
Il est probable que l’insuffisance cardiaque droite sévère de ces patients soit causée par une lésion des artères pulmonaires due à l’inhalation de fumée de cigarette. L’utilisation de médicaments spécifiques à l’HTP est probablement beaucoup moins efficace chez les patients souffrant d’une forme d’hypertension pulmonaire plus probablement associée au tabagisme que chez les patients IPAH classiques.
Étant donné que ces patients ont été exclus ou sous-représentés dans les études d’enregistrement des médicaments spécifiques à l’HTP menées jusqu’à présent en raison des critères d’inclusion et d’exclusion, il est urgent de mener des études prospectives, randomisées et contrôlées par placebo, à la fois pour caractériser plus précisément ces patients et pour clarifier les options thérapeutiques et la place des médicaments spécifiques à l’HTP.
Messages Take-Home
- Le groupe de patients atteints de l’IPAH est plus hétérogène qu’on ne le pensait jusqu’à présent.
- Les patients qui répondent aux critères de l’IPAH mais qui présentent des signes d’HTP associée au tabagisme devraient à l’avenir faire l’objet d’une plus grande attention scientifique afin de déterminer à quel groupe d’HTP ces patients appartiennent et quelles sont les options thérapeutiques efficaces disponibles.
- Des études thérapeutiques prospectives, contrôlées par placebo et randomisées sont nécessaires de toute urgence pour clarifier les options thérapeutiques d’une HTP probablement associée au tabagisme.
Littérature :
- Lewis RA, Thompson AAR, Billings CG, et al : Mild parenchymal lung disease and/or low diffusion capacity impacts survival and treatment response in patients diagnosed with idiopathic pulmonary arterial hypertension. European Respiratory Journal ; doi : 10.1183/13993003.00041-2020.
- Hoeper MM, Pausch C, Grünig E, et al : Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. Journal of Heart and Lung Transplantation 2020 ; 39 : 1435-1444 ; doi : 10.1016/j.healun.2020.09.011.
- Trip P, Nossent EJ, De Man FS, et al : Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension : patient characteristics and treatment responses. European Respiratory Journal ; doi : 10.1183/09031936.00184412.
- Chandra S, Shah SJ, Thenappan T, et al : Capacité de diffusion du monoxyde de carbone et mortalité dans l’hypertension artérielle pulmonaire. Journal of Heart and Lung Transplantation 2010 ; 29 : 181-187 ; doi : 10.1016/J.HEALUN.2009.07.005.
- Olsson KM, Fuge J, Meyer K, et al : More on idiopathic pulmonary arterial hypertension with a low diffusing capacity. European Respiratory Journal 2017 ; 50 ; doi : 10.1183/13993003.00354-2017.
- Hoeper MM, Apitz C, Grünig E, et al : Traitement ciblé de l’hypertension artérielle pulmonaire. DMW 2016 ; S33-S41.
- Hoeper MM, Apitz C, Grünig E, et al : Targeted therapy of pulmonary arterial hypertension : Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 2018 ; 272 : 37-45 ; doi : 10.1016/j.ijcard.2018.08.082.
- Trip P, Nossent EJ, De Man FS, et al : Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension : Patient characteristics and treatment responses. European Respiratory Journal 2013 ; 42 : 1575-1585 ; doi : 10.1183/09031936.00184412.
- Hoeper MM, Dwivedi K, Pausch C, et al : Phenotyping of idiopathic pulmonary arterial hypertension : a registry analysis. Lancet Respir
Med 2022 ; 10(10) : 937-948 ; doi : 10.1016/S2213-2600(22)00097-2.
COIs :
- Halank : Honoraires pour les activités de conférence et de conseil
d’AstraZeneca, de Janssen et de MSD. Frais de déplacement de Janssen.
Tout ce qui n’est pas en rapport avec la revue actuelle - Heberling : honoraires de conférencier et de consultant et frais de déplacement de Janssen-Cillag et MSD.
- Kolditz : aucune en rapport avec ce travail
- Koschel : aucun en rapport avec ce travail
InFo PNEUMOLOGIE & ALLERGOLOGIE 2023 ; 5(3) : 6-11









