La présence d’une DLI chez les patients atteints de rhumatisme limite le pronostic et la survie ainsi que la qualité de vie. Le diagnostic précoce est d’une grande importance, car il permet de commencer le traitement à temps et de prévenir la progression, qui a un impact sur le pronostic. Les exacerbations aiguës de l’ILD peuvent également compliquer l’évolution de la maladie. Ils sont soit associés à la progression de la maladie, soit déclenchés secondairement par des infections.
Les pneumopathies interstitielles (PID) se rencontrent principalement dans les collagénoses, notamment la sclérose systémique (SSc), les myopathies auto-immunes, le syndrome de Sjögren, le lupus érythémateux disséminé (LED), mais aussi la polyarthrite rhumatoïde (PR). Lors de l’évaluation clinique rhumatologique, la personne concernée doit toujours être interrogée sur la présence éventuelle d’une limitation respiratoire. La présence d’une DLI chez les patients atteints de rhumatisme limite le pronostic, la survie et la qualité de vie [1]. Le diagnostic précoce est d’une grande importance, car il permet de commencer le traitement à temps et de prévenir la progression, qui a un impact sur le pronostic. La progression de la DLI est fréquente et s’observe en particulier chez les patients atteints de SSc et de PR dans des proportions importantes ; des critères de détermination de la progression ont été récemment publiés [2]. Les exacerbations aiguës de l’ILD peuvent également compliquer l’évolution de la maladie. Ils sont soit associés à la progression de la maladie, soit déclenchés secondairement par des infections.
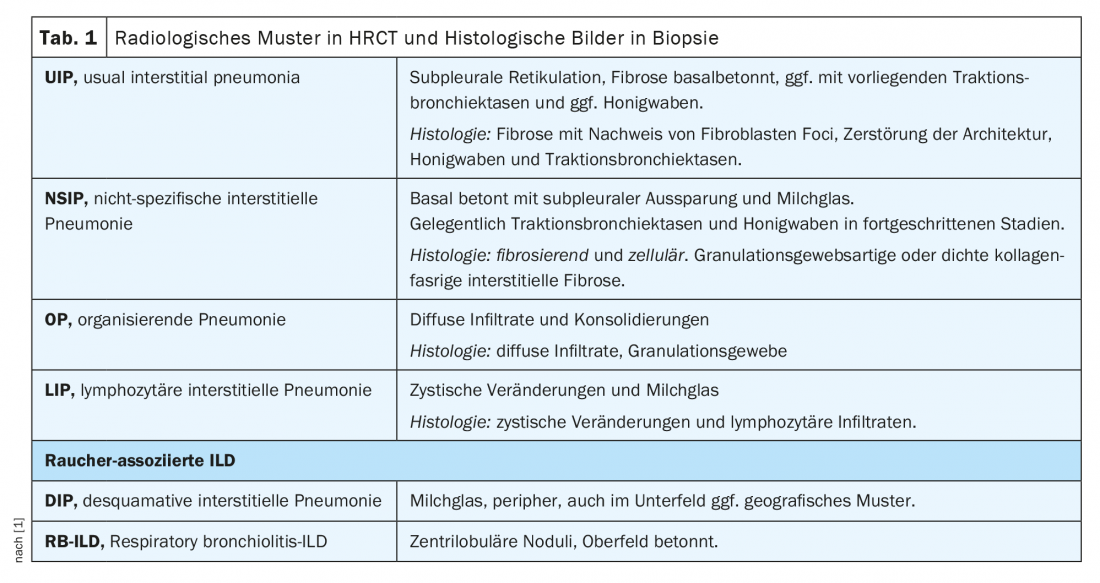
Tableau ILD
Dans le cadre du conseil d’administration de l’ILD, les cas sont discutés de manière interdisciplinaire sur la base des paramètres suivants : symptômes dominants, antécédents, y compris les facteurs de risque. Expositions et toxines, immunosérologie, limitation de la fonction pulmonaire dans la pléthysmographie corporelle (en particulier les maladies respiratoires). FVC, TLC, FEV1) et capacité de diffusion (DLCO), imagerie par tomodensitométrie thoracique à haute résolution (HRCT) ainsi que résultats du diagnostic invasif par bronchoscopie, y compris Microbiologie, résultats du lavage broncho-alvéolaire (LBA) et résultats histologiques des biopsies (tableau 1). Les patients atteints de rhumatisme peuvent développer une DLI non seulement à cause de la maladie rhumatismale, mais aussi à cause de toxines, de médicaments ou d’expositions (surtout en cas de LBA lymphocytaire), etc. (Fig. 1). Il convient de noter que le prélèvement de biopsies est rarement indiqué de manière stricte dans les maladies rhumatismales confirmées, mais il sert à faciliter la délimitation des diagnostics différentiels, en particulier dans les vascularites et surtout dans la coexistence de maladies tumorales et d’autres maladies.
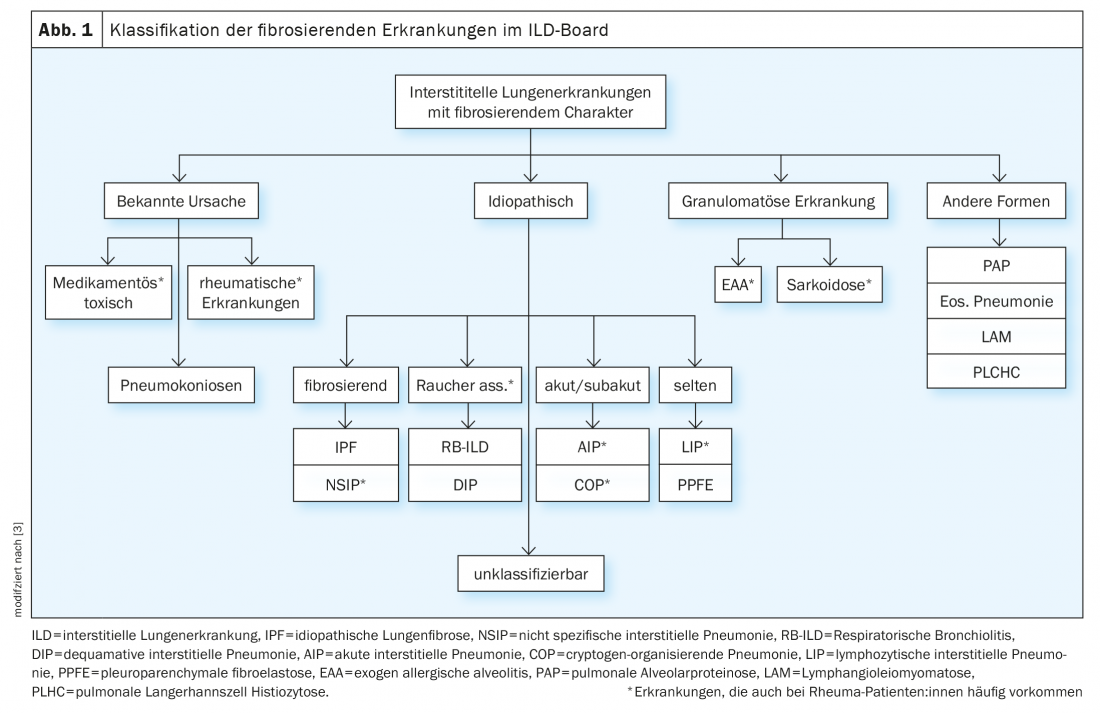
Maladies rhumatismales inflammatoires avec implication possible dans le DLI
PR-ILD : La PR est la maladie rhumatismale la plus fréquente. La présence d’arthrites, en particulier dans les petites articulations, ainsi que de facteurs rhumatoïdes et d’anticorps contre les peptides citrullinés cycliques élevés (ce que l’on appelle la séropositivité) simplifient le diagnostic et constituent donc des éléments d’orientation [4]. La manifestation extra-articulaire la plus fréquente de la PR est la DLI, qui se manifeste chez 25 à 60% des patients atteints de PR à l’HRCT et est responsable de 10 à 20% des décès dus à la PR. Chez environ 10% des personnes atteintes, la PR-ILD devient cliniquement significative. Dans 10% des cas, l’atteinte pulmonaire précède même le développement de l’arthrite [5]. Les facteurs de risque de développer une ILD incluent le sexe masculin, le tabagisme et la séropositivité. L’imagerie révèle le plus souvent un motif UIP. Des mutations comme celles du promoteur du gène MUC5B sont décrites dans la littérature. Le tabagisme peut en outre entraîner une aggravation de la situation pulmonaire. La présence d’un trouble ventilatoire combiné (obstructif et restrictif), dans le sens d’un CPFE (combined pulmonary fibrosis and emphysema) , est associée à un pronostic dramatiquement plus mauvais. Le diagnostic différentiel à envisager est celui d’une DLI médicamenteuse toxique sous immunosuppression. La DLI associée au méthotrexate (MTX) est toutefois extrêmement rare (estimée à 0,1%) ; un effet protecteur du MTX a même été décrit.
Le MTX ne doit donc pas être arrêté en cas de PR-ILD. Le traitement de la PR-ILD passe en premier lieu par l’optimisation de l’immunosuppression. Les études randomisées et contrôlées pour le traitement de la PR-ILD font défaut. Le traitement par abatacept et rituximab des patients atteints de PR et de DLI concomitante est le mieux documenté [6]. Les résultats de l’étude APRIL (abatacept dans la PR-ILD) sont encore attendus. De plus, l’efficacité des inhibiteurs de JAK ou des bloqueurs d’IL-6 dans le DLI de la PR a été rapportée récemment dans des séries de cas [7]. En cas d’évolution progressive (aperçu 1), il est recommandé d’initier un traitement antifibrotique. Pour cela, le nintédanib a été approuvé dans l’UE sur la base des résultats de l’étude INBUILD [8]. Le pronostic des patients atteints de PR-ILD est globalement très limité, avec une durée de survie moyenne d’environ trois ans [1].

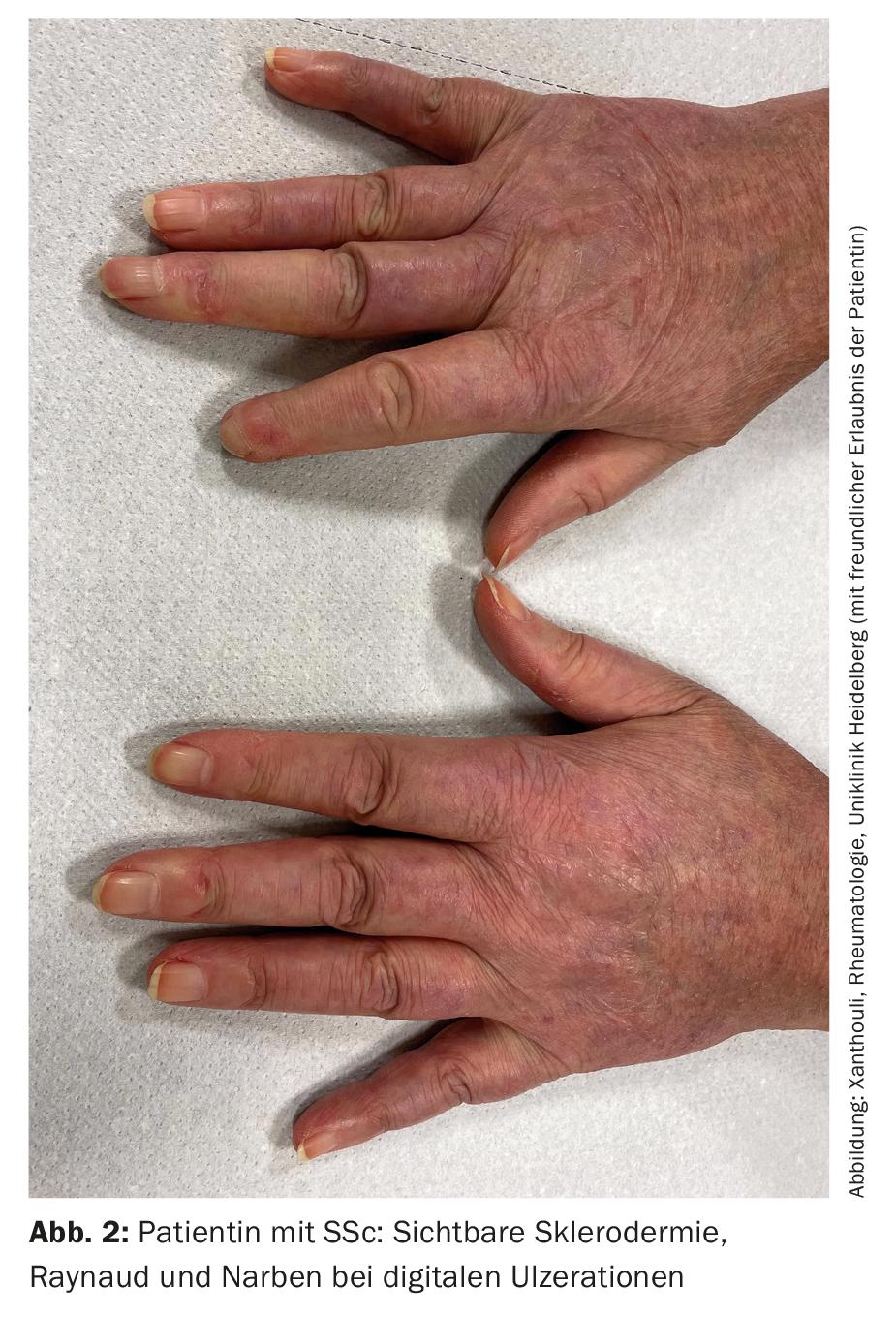
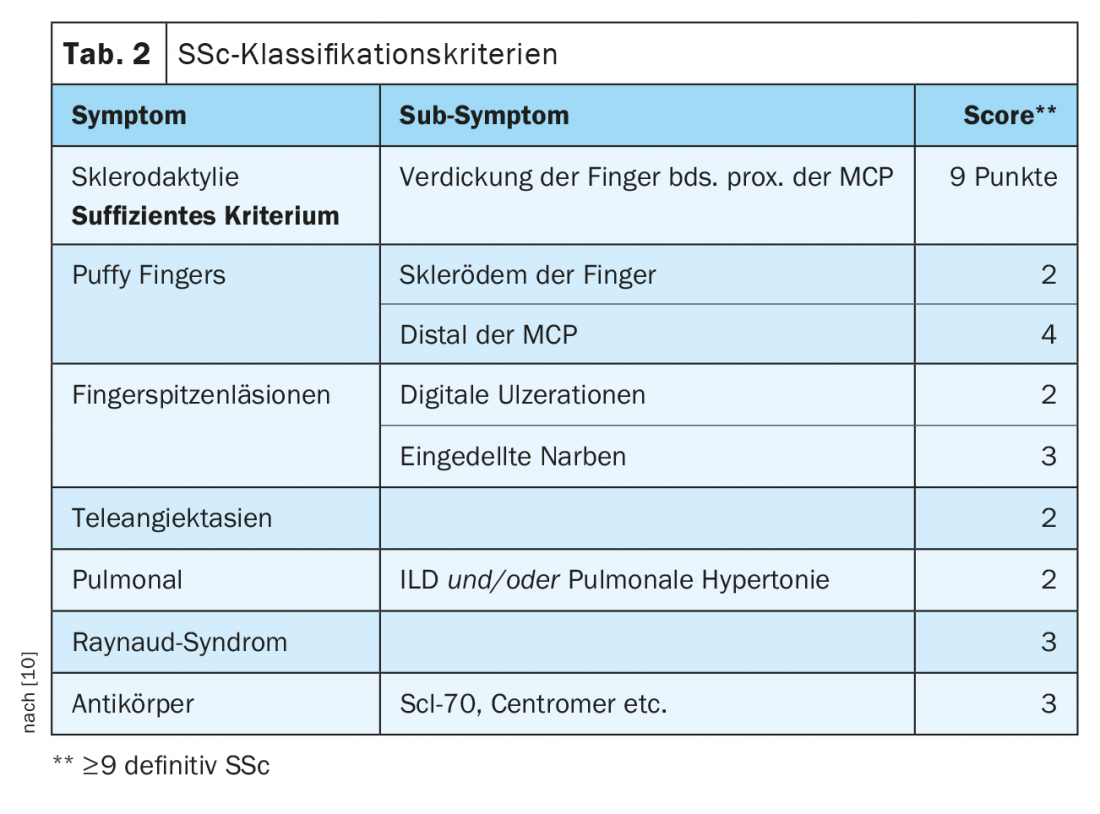
SSc-ILD : environ 50 à 85% des patients atteints de SSc présentent des lésions interstitielles pulmonaires dès le diagnostic initial. Dans 20 à 30% des cas, elle est progressive au cours de l’évolution. L’ILD est actuellement la principale cause de décès chez les patients atteints de SSc, avec un taux de mortalité à 10 ans d’environ 40% [1]. Un diagnostic précoce et correct ainsi que la reconnaissance d’une ILD ont une incidence sur le pronostic [9]. Des critères de classification ont été établis pour faciliter l’établissement du diagnostic (tableau 2) [10]. La sclérodactylie, les ulcérations digitales, la symptomatologie de Raynaud et la microstomie sont des signes cliniques qui doivent immédiatement faire penser à une SSc (Fig. 2). L’apparition et surtout la progression de la maladie ont lieu principalement au cours des trois premières années de la maladie. Souvent, l’ILD est asymptomatique chez les SSc. Il est recommandé de procéder à un dépistage par HRCT lors du diagnostic initial de la SSc. Des examens fonctionnels pulmonaires (body-pléthysmographie et mesures de la capacité de diffusion) sont nécessaires tous les trimestres ou semestres. Les facteurs de risque de développer une ILD sont le sexe masculin, la présence d’une sclérose systémique cutanée diffuse, la détection d’auto-anticorps anti-Scl-70 et l’origine afro-américaine [11]. Le reflux, l’atteinte cutanée diffuse (et donc un score de peau modifié de Rodnan plus élevé) et le sexe masculin sont des facteurs de risque de progression rapide de la DLI. En TDM, on observe le plus souvent un schéma NSIP, et plus rarement un schéma UIP.
Le traitement est principalement immunomodulateur et, en complément, antifibrotique [1]. Sur la base des résultats des études SLS I et II, un traitement par cyclophosphamide ou mycophénolate mofétil peut être initié dans le cas de la SSc-ILD [12,13]. Dans l’étude FocuSSed, le tocilizumab (anticorps dirigé contre le récepteur IL-6) a permis d’améliorer la CVF dans le phénotype inflammatoire de la SSc. Le tocilizumab a donc reçu l’approbation de la FDA pour l’indication SSc-ILD, mais il est encore utilisé hors étiquette en Europe [14]. Pour le rituximab (également encore off label), on dispose actuellement de bonnes données pour le traitement de la SSc-ILD provenant de l’étude DESIRES [15]; les résultats définitifs de l’étude RECITAL sont encore attendus. L’étude SCENCIS est la première étude contrôlée, randomisée, en double aveugle et de grande envergure à évaluer l’efficacité du traitement antifibrotique par nintédanib chez les patients atteints de SSc-ILD [16]. Le nintédanib est approuvé par la FDA pour le traitement de la SSc-ILD, mais il n’agit que pour prévenir la progression de l’atteinte pulmonaire et ne convient pas pour le traitement d’autres manifestations de la SSc.
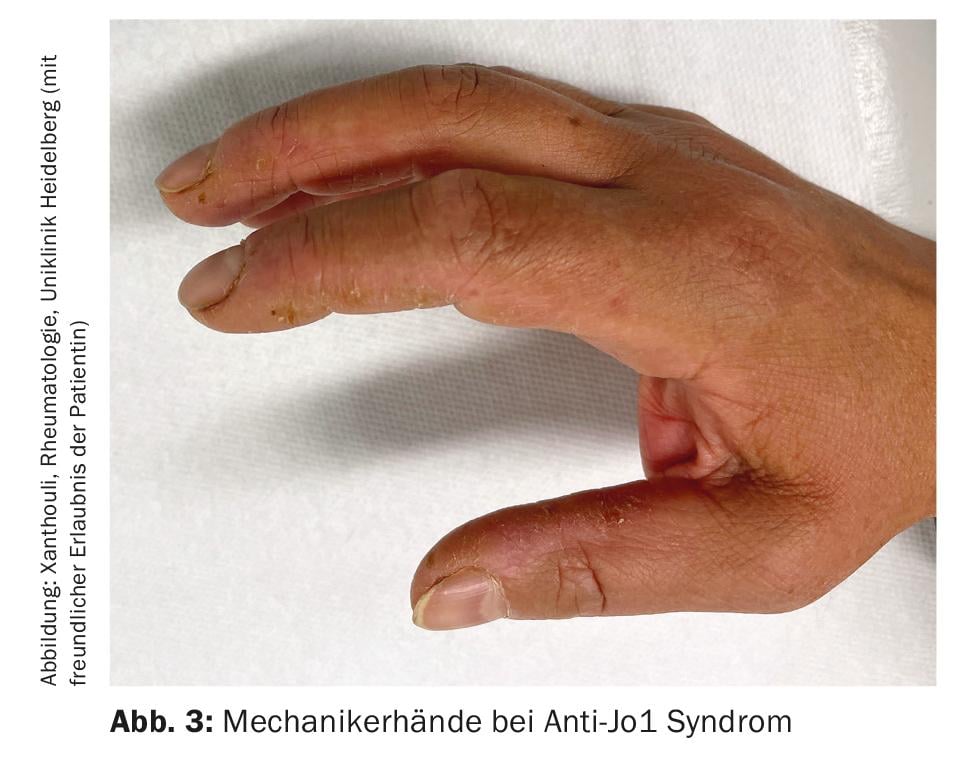
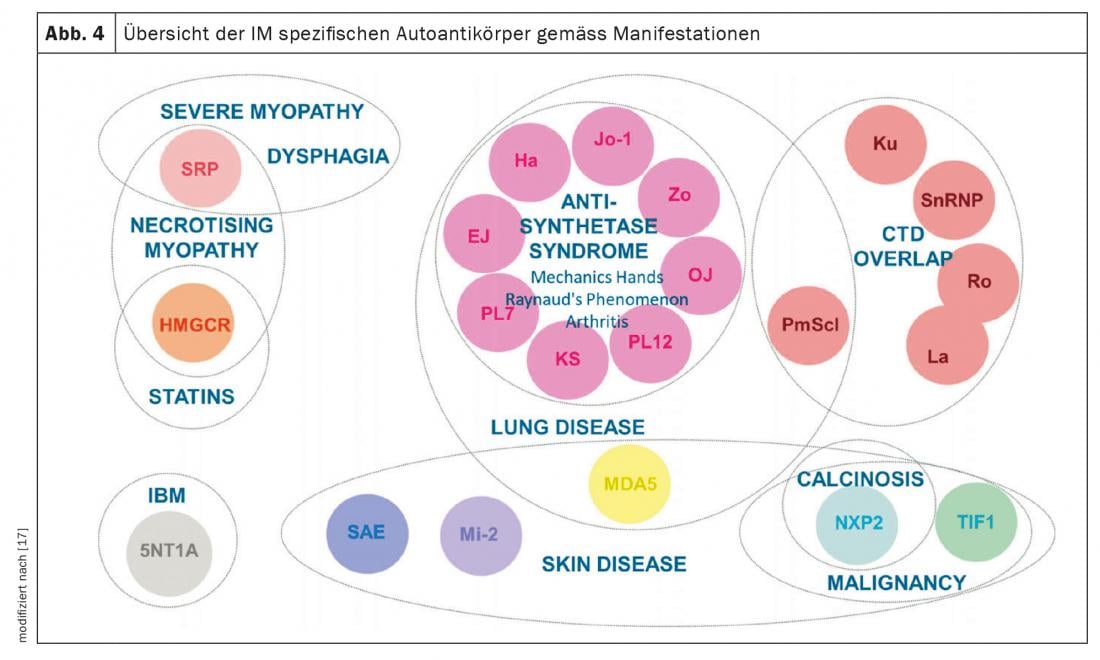
Myosites inflammatoires (MI) : les MI comprennent le syndrome de l’antisynthétase (SA), la dermato- et la polymyosite, la myosite à corps inclus et les syndromes de chevauchement avec d’autres collagénoses. Elles se manifestent avec ou sans atteinte musculaire (myosite). Les patients présentant une atteinte musculaire ont des taux sanguins de muscle exceptionnellement élevés (CK, créatine kinase) et présentent des signaux pathologiques à l’IRM musculaire et à l’examen neurophysiologique. Les autres manifestations cliniques évocatrices de la MI sont les mains de mécanicien (figure 3), surtout en cas de syndrome des anti-synthétases, les arthrites, un exanthème héliotrope en cas de dermatomyosite et un phénomène de Raynaud. Jusqu’à 70% des patients atteints d’ASA développent une DLI. De plus, chez un tiers des personnes atteintes de MI, la DLI est la première manifestation de la maladie [7]. Les auto-anticorps les plus fréquents dans les ASA sont les anticorps anti-Jo-1, mais d’autres anticorps sont connus, par exemple contre PL12, PL7, Mi2, etc. et doivent être recherchés en cas de symptômes typiques. La détection d’anticorpsMDA5 (melanoma differentiation accociated protein 5)en cas d’atteinte pulmonaire indéterminée indique une évolution rapidement progressive, réfractaire au traitement et associée à une mortalité plus élevée, souvent sans myosite. Il convient de noter l’association entre les tumeurs malignes et la MI. Elle est particulièrement élevée en cas de détection d’anticorps TIFγ et NXP2. Dans ce cas, une tumeur maligne est présente dans 30% des cas, ce qui rend indispensable un dépistage approfondi de la tumeur (Fig. 4) [17].
Le traitement est principalement basé sur l’administration de stéroïdes à haute dose, avec une bonne réponse, en particulier chez les jeunes patients, les patients opérés, les taux élevés de CK et les infiltrations de verre dépoli à l’HRCT. Dans les cas graves, l’initiation d’un traitement pulsé par cyclophosphamide doit être envisagée. Le mycophénolate, le rituximab, le tacrolimus, l’abatacept et les inhibiteurs de JAK restent des traitements off label, bien qu’ils soient soutenus par de bons résultats d’études rétrospectives [18]. Il convient également de noter qu’il existe une autorisation de mise sur le marché pour l’administration d’immunoglobulines intraveineuses dans la myosite dans l’UE [19]. Chez les patients présentant des anticorps anti-Jo-1 et des anticorps anti-Ro-52, le rituximab a récemment montré une bonne réponse en termes d’implication dans la DLI, ce qui justifie de l’envisager comme option thérapeutique [20].
LED : les multiples manifestations pulmonaires du LED entraînent souvent un retard de diagnostic. Jusqu’à 12% des personnes atteintes pourraient développer une DLI au cours de l’évolution. Une manifestation particulièrement dangereuse du LED est le syndrome hémorragique pulmonaire, qui se caractérise par une hémoptysie sévère et une détérioration respiratoire rapide. En revanche, une dyspnée sévère, un trouble ventilatoire restrictif avec apraxie diaphragmatique sans présence d’ILD font suspecter la présence d’un Shrinking Lung Syndrome difficile à traiter . La demande d’une échographie thoracique, la mesure de la force des muscles respiratoires et la spiro-ergométrie peuvent être orientées en conséquence. Le traitement de la DLI dans le LED passe en premier lieu par l’intensification de l’immunosuppression. Les antifibrotiques pourraient être utilisés en cas de progression – dans de rares cas ; une approbation pour le nintédanib dans l’UE [21].
Syndrome de Sjögren : bien que la majorité des patients atteints du syndrome de Sjögren n’expriment guère de symptômes pulmonaires, le diagnostic de la fonction pulmonaire révèle dans certains cas un trouble de la ventilation restrictive. La présence d’une symptomatologie siccative objectivable avec des anticorps anti-SSA positifs est déterminante pour le diagnostic de la maladie sous-jacente [22]. Dans 15% des cas, l’HRCT présente un schéma LIP avec des modifications kystiques bipulmonaires diffuses avec verre dépoli. La biopsie révèle un infiltrat lymphocytaire. En cas de masse intrathoracique inexpliquée dans le cadre d’un syndrome de Sjögren primaire, il convient d’envisager un diagnostic différentiel avec un lymphome. Le diagnostic précoce d’une DLI associée au syndrome de Sjögren est important pour le pronostic avec une survie à 5 ans de 88,5% [23].
Pneumonie interstitielle à caractéristiques auto-immunes (IPAF) : Les patients atteints de la maladie de Lyme et présentant des anticorps anti-nucléaires (ANA) anormaux sont souvent consultés en rhumatologie. Dans 10 à 20 % des cas, il s’agit de découvertes fortuites dans le cadre du diagnostic de dépistage de l’ILD. En HRCT, il existe le plus souvent un schéma NSIP et les personnes concernées bénéficient nettement des stéroïdes. Cependant, d’autres symptômes évocateurs d’une maladie rhumatismale inflammatoire sont rarement présents. Dans ce cas, un suivi combiné chez le pneumologue et le rhumatologue est nécessaire pour identifier à temps une manifestation tardive d’une maladie rhumatismale inflammatoire [7].
Autres maladies
Les vascularites : Chez les patients atteints de granulomatose avec polyangéite (GPA), des granumolomes intrapulmonaires peuvent souvent être détectés. Une ILD peut se développer dans de rares cas. En revanche, en cas de détection d’anticorps anti-myélopéroxydase (MPO-ANCA), une DLI présentant principalement un schéma UIP est souvent décrite. Dans ce cas, il convient d’effectuer un diagnostic complémentaire concernant les autres manifestations organiques.
Collagénose mixte : la fréquence de l’ILD est estimée à 47-78%, ce qui est très variable. Les personnes concernées ont des anticorps ANA et U1-RNP positifs. En HRCT, il peut y avoir soit un motif NSIP, soit un motif UIP, soit des infiltrats dans le sens d’une opération. Le traitement repose principalement sur des substances immunomodulatrices [7].
Autre traitement non médicamenteux et prophylactique
Tous les patients atteints de maladies rhumatismales doivent recevoir les vaccins recommandés, notamment contre le COVID-19, la grippe et le pneumocoque, et les mettre à jour régulièrement. Dans les stades avancés de l’ILD, une défaillance respiratoire peut survenir. Une substitution d’oxygène est souvent nécessaire au cours de l’évolution, en commençant par l’effort, puis, le cas échéant, par le repos. Pour cela, une titration est généralement effectuée pour déterminer la dose. Les mesures de réhabilitation telles que le sport pulmonaire et rhumatologique ainsi que les mesures de réhabilitation stationnaires ou ambulatoires améliorent la situation clinique et l’absorption d’oxygène des personnes atteintes de maladies rhumatismales inflammatoires et sont toujours recommandées. Si les possibilités thérapeutiques sont épuisées, il convient d’envisager une inscription sur la liste des transplantations pulmonaires. Il n’existe pas de directives claires concernant le moment exact de la présentation au référencement. Dans le cas de la SSc, l’autogreffe de cellules souches prend de plus en plus d’importance, en particulier dans les cas d’évolution clinique sévère [24]. Si la transplantation n’est pas envisageable, la mise en place de mesures palliatives et de soulagement des symptômes doit être envisagée [25].
Résumé
Les maladies rhumatismales peuvent se manifester dans le parenchyme pulmonaire, où elles entraînent notamment le développement de pneumopathies interstitielles (PID). Les manifestations pulmonaires peuvent précéder la maladie rhumatismale sous-jacente dans environ 10% des cas. C’est pourquoi des rhumatologues sont impliqués dans le cadre du conseil interdisciplinaire ILD afin de contribuer au diagnostic différentiel et à la définition de l’algorithme thérapeutique. Le pronostic de la DLI rhumatismale est limité, mais une collaboration entre les rhumatologues et les pneumologues peut améliorer considérablement la qualité des soins. Bien que la pathogenèse de l’apparition de la DLI reste obscure, le développement des traitements médicamenteux de la DLI rhumatismale progresse. Le rhumatologue et le pneumologue doivent tous deux posséder des connaissances approfondies et être à l’aise avec les substances immunomodulatrices et antifibrotiques.
Messages Take-Home
- Les maladies rhumatismales inflammatoires peuvent se manifester dans le parenchyme pulmonaire.
- Une coopération étroite entre les pneumologues et les rhumatologues est nécessaire pour établir le diagnostic et le traitement.
- Le traitement est principalement immunosuppresseur.
- Depuis 2020, l’agent antifibrotique nintedanib a été approuvé pour le traitement de la SSc-ILD sur la base des résultats de l’étude SENSCIS. Le médicament est également utilisé dans le traitement de l’ILD progressive (étude INBUILD).
Littérature :
- Wijsenbeek M, Cottin V : Spectrum of Fibrotic Lung Diseases. N Engl J Med 2020 ; 383 : 958-968 ; doi : 10.1056/NEJMra2005230.
- Raghu G, Remy-Jardin M, Richeldi L et al : Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults : An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2022 ; 205 : e18-e47 ; doi : 10.1164/rccm.202202-0399ST.
- Kreuter M, Ladner UM, Costabel U, et al : The Diagnosis and Treatment of Pulmonary Fibrosis. Dtsch Arztebl Int 2021 ; 118 ; doi : 10.3238/arztebl.m2021.0018.
- Aletaha D, Neogi T, Silman AJ, et al : 2010 rheumatoid arthritis classification criteria : an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2010 ; 69 : 1580-1588 ; doi : 10.1136/ard.2010.138461.
- Duarte AC, Porter JC, Leandro MJ : Les poumons dans une cohorte de patients atteints d’arthrite rhumatoïde – un aperçu des différents types d’implication et de traitement. Rheumatology (Oxford) 2019 ; 58 : 2031-2038 ; doi : 10.1093/rheumatology/kez177.
- Mena-Vázquez N, Rojas-Gimenez M, Romero-Barco CM, et al : Predictors of Progression and Mortality in Patients with Prevalent Rheumatoid Arthritis and Interstitial Lung Disease : A Prospective Cohort Study. J Clin Med 2021 ; 10(4) : 874.
- Bastian HKA : Poumons – Pneumopathies interstitielles. Akt Rheumatol 2021 ; 46 : 544-551.
- Flaherty KR, Wells AU, Cottin V, et al : Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med 2019 ; 381 : 1718-1727 ; doi : 10.1056/NEJMoa1908681.
- Xanthouli P, Hermann W, Hunzelmann N, et al : Pneumopathies interstitielles associées à la sclérodermie. Le Pneumologue 2018 ; 15 : 383-395.
- van den Hoogen F, Khanna D, Fransen J, et al : 2013 classification criteria for systemic sclerosis : an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis 2013 ; 72 : 1747-1755 ; doi : 10.1136/annrheumdis-2013-204424.
- Distler O, Volkmann ER, Hoffmann-Vold AM, et al : Current and future perspectives on management of systemic sclerosis-associated interstitial lung disease. Expert Rev Clin Immunol 2019 ; 15 : 1009-1017 ; doi : 10.1080/1744666X.2020.1668269.
- Tashkin DP, Elashoff R, Clements PJ, et al : Cyclophosphamide versus placebo dans la maladie pulmonaire sclérodermique. N Engl J Med 2006 ; 354 : 2655-2666 ; doi : 10.1056/NEJMoa055120.
- Tashkin DP, Roth MD, Clements PJ, et al : Mycophénolate mofetil versus cyclophosphamide oral dans la maladie pulmonaire interstitielle liée à la sclérodermie (SLS II) : une étude randomisée contrôlée, en double aveugle, en groupes parallèles. Lancet Respir Med 2016 ; 4 : 708-719 ; doi : 10.1016/S2213-2600(16)30152-7.
- Khanna D, Lin CJF, Furst DE, et al : Tocilizumab in systemic sclerosis : a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 2020 ; 8(10) : 963-974.
- Ebata S, Yoshizaki A, Oba K, et al. : Safety and efficacy of rituximab in systemic sclerosis (DESIRES) : a double-blind, investigator-initiated, randomised, placebo-controlled trial. The Lancet Rheumatology 2021 ; 3 : E489-E497 ; doi : 10.1016/S2665-9913(21)00107-7.
- Distler O, Highland KB, Gahlemann M, et al : Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med 2019 ; 380(26) : 2518-2528.
- Betteridge Z, McHugh N : Autoantibodies spécifiques à la myosite : un outil important pour aider au diagnostic de la myosite. J Intern Med 2016 ; 280 : 8-23 ; doi : 10.1111/joim.12451.
- Mehta P, Aggarwal R, Porter JC, et al : Management of interstitial lung disease (ILD) in myositis syndromes : A practical guide for clinicians. Best Pract Res Clin Rheumatol 2022 ; 101769 ; doi : 10.1016/j.berh.2022.101769.
- Aggarwal R, Charles-Schoeman C, Schessl J, et al. : Étude prospective, en double aveugle, randomisée, contrôlée par placebo de phase III évaluant l’efficacité et la sécurité de l’octagam 10% chez les patients atteints de dermatomyosite (“Étude ProDERM”). Medicine (Baltimore) 2021 ; 100 : e23677 ; doi : 10.1097/MD.0000000000023677.
- Bauhammer J, Blank N, Max R, et al : Rituximab dans le traitement du syndrome des anti-synthétases associées à Jo1 : positivité anti-Ro52 comme marqueur de sévérité et de réponse au traitement. J Rheumatol 2016 ; 43 : 1566-1574 ; doi : 10.3899/jrheum.150844.
- Medlin JL, Hansen KE, McCoy SS, et al : Manifestations pulmonaires dans le lupus érythémateux disséminé tardif versus précoce : A systematic review and meta-analysis. Semin Arthritis Rheum 2018 ; 48 : 198-204 ; doi : 10.1016/j.semarthrit.2018.01.010.
- Shiboski CH, Shiboski SC, Seror R, et al : 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjogren’s Syndrome : A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol 2017 ; 69 : 35-45 ; doi : 10.1002/art.39859.
- Gao H, Zhang XW, He J, et al : Prévalence, facteurs de risque et pronostic de la pneumopathie interstitielle dans une large cohorte de patients chinois atteints du syndrome de Sjogren primaire : une étude cas-témoins. Medicine (Baltimore) 2018 ; 97 : e11003 ; doi : 10.1097/MD.0000000000011003.
- Farge D, Ait Abdallah N, Marjanovic Z, et al : Transplantation de cellules souches autologues dans la sclérodermie. Presse Med 2021 ; 50 : 104065 ; doi : 10.1016/j.lpm.2021.104065.
- Kreuter M, Bendstrup E, Russell AM, et al : Soins palliatifs dans les maladies pulmonaires interstitielles : vivre bien. Lancet Respir Med 2017 ; 5 : 968-980 ; doi : 10.1016/S2213-2600(17)30383-1.
PRATIQUE DU MÉDECIN DE FAMILLE 2022 ; 17(9) : 4-9