Les neuropathies à médiation immunitaire varient dans leur présentation clinique, leur évolution et leur pathogenèse. Ces dernières années, des auto-anticorps dirigés contre les protéines nodales et paranodales de l’anneau de Ranvier ont été détectés. Les auto-anticorps peuvent provoquer des lésions axonales et entraîner une démyélinisation.
Les neuropathies à médiation immunitaire varient en termes de présentation clinique, d’évolution et de pathogenèse et représentent environ 9% de toutes les polyneuropathies [1]. Au cours des dernières années, des auto-anticorps dirigés contre les protéines nodales et paranodales de l’anneau de Ranvier ont été détectés. Des auto-anticorps dirigés contre la neurofascine (NF), la contactine 1 (CNTN1) ou la protéine 1 associée à la contactine (CASPR1) ont été détectés chez 10% des patients atteints de polyneuropathie inflammatoire démyélinisante chronique (PIDC) [2]. Les auto-anticorps peuvent provoquer des lésions axonales et entraîner une démyélinisation [4]. Les patients séropositifs atteints de PIDC se distinguent des patients séronégatifs par leur phénotype clinique, leurs résultats électrophysiologiques et leur réponse au traitement standard [2].
La détection d’auto-anticorps spécifiques nodaux et paranodaux peut aider à trouver des traitements efficaces. Outre la PIDC, des auto-anticorps associés à la maladie sont également connus dans d’autres neuropathies à médiation immunitaire, comme le syndrome de Guillain-Barré (SGB), dont les sous-types sont la polyneuropathie inflammatoire démyélinisante aiguë (AIDP), la neuropathie axonale motrice aiguë (AMAN) et la neuropathie axonale motrice et sensitive aiguë (AMSAN), le syndrome de Miller-Fisher (MFS), la neuropathie motrice multifocale (MMN), la neuropathie anti-MAG MGUS (neuropathie paraprotéinémique), la neuronopathie sensitive des maladies immunitaires systémiques et les polyneuropathies paranéoplasiques.
Physiopathologie et modifications électrophysiologiques des nodo- et paranodopathies
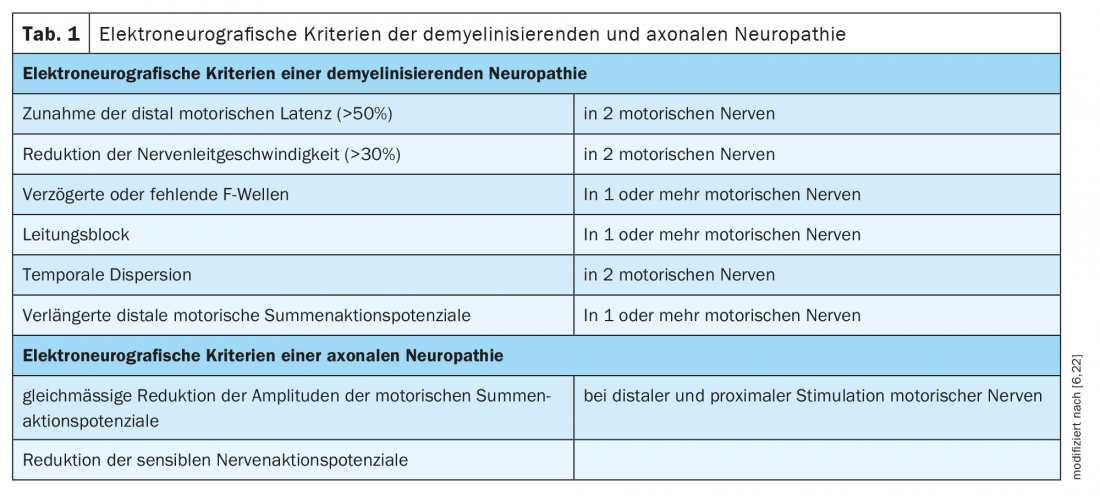
Les polyneuropathies sont traditionnellement divisées en neuropathies démyélinisantes et axonales selon des critères électrophysiologiques (tableau 1). La pathologie sous-jacente est attendue en conséquence au niveau de la cellule de myéline/chwan ou de l’axone. Les protéines de la région nodale ou paranodale de l’anneau de Ranvier sont des jonctions entre la myéline/les microvillosités de la cellule de Schwan et l’axone (Fig. 1). Dans les neuropathies dues à des auto-anticorps dirigés contre les protéines nodales et paranodales, il n’est pas possible d’attribuer clairement un modèle démyélinisant ou axonal en raison du mécanisme pathologique sous-jacent. Le terme de nodo- et paranodopathies a été proposé par Uncini, Susuki et Yuki en 2013 [8,9].
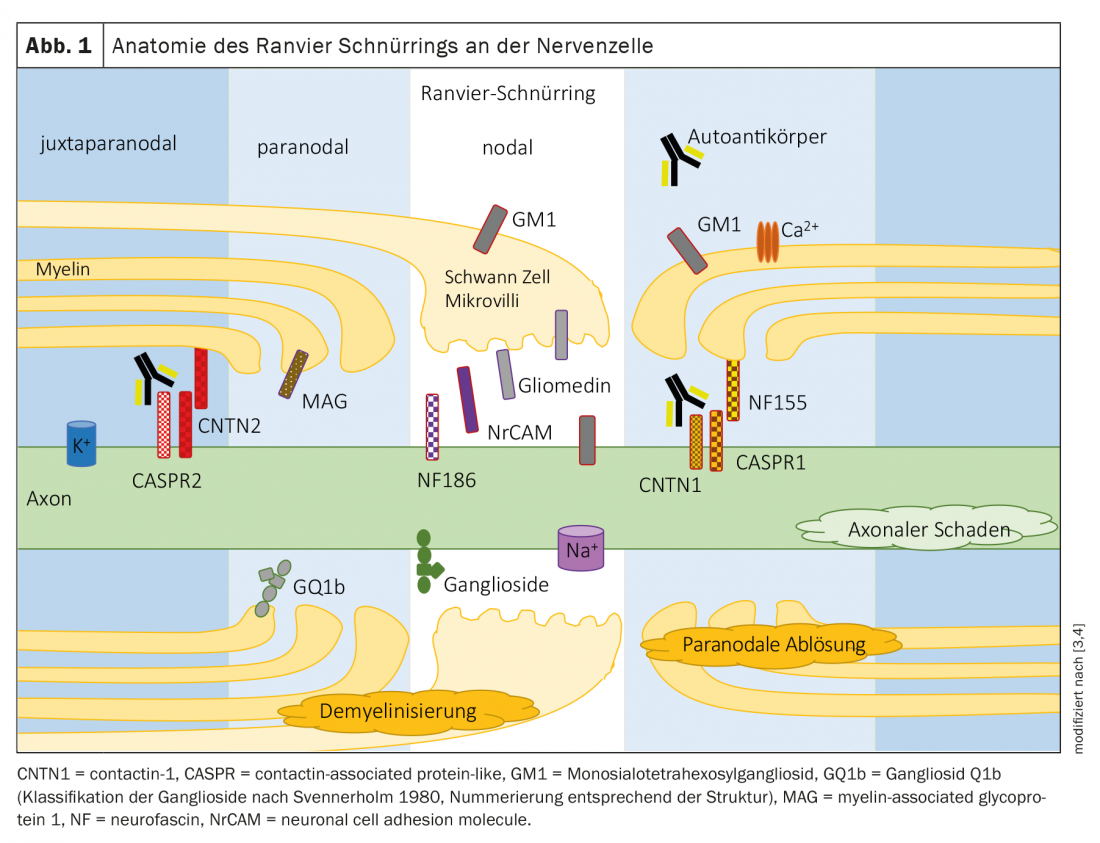
Les caractéristiques d’une nodo- et d’une paranodopathie sont les suivantes
- Les anticorps dirigés contre les protéines nodales et paranodales ont des étiologies différentes, mais ils entraînent tous un dysfonctionnement de la région nodale de l’anneau de Ranvier.
- Continuum pathologique allant du bloc de conduction transitoire à la dégénérescence axonale.
- Bloc de conduction dû au détachement de la myéline de l’axone, à l’élargissement de la région nodale ou à un dysfonctionnement/une perturbation des canaux sodiques (densité élevée dans la région nodale pour la transmission saltatoire du stimulus) avec une polarisation anormale segmentaire de l’axolemme.
- Le bloc de conduction peut être rapidement réversible sans apparition d’une dispersion temporelle nette, le bloc de conduction peut également être maintenu.
- La dégénérescence axonale dépend de la maladie spécifique et de sa sévérité, éventuellement suivie d’un bloc de conduction.
- Le diagnostic peut être établi par des examens électrophysiologiques répétés :
- a. lors d’une maladie aiguë, en cas de bloc de conduction rapidement réversible ou de diminution de la vitesse de conduction nerveuse sans dispersion temporale marquée ou de progression d’un bloc de conduction vers une dégénérescence axonale ;
- b. dans les maladies chroniques, en cas de bloc de conduction persistant et de signes supplémentaires de dégénérescence axonale.
L’utilisation du terme de nodo- et paranodopathies met l’accent sur la localisation de l’atteinte nerveuse primaire et aide à mieux situer la neuropathie segmentaire démyélinisante, le paradoxe de l’atteinte axonale réversible et le bon pronostic, malgré des anomalies axonales à l’électrophysiologie [9].
Auto-anticorps dans la polyneuropathie inflammatoire démyélinisante chronique (PIDC)
Localisation : Dans la région paranodale, le complexe de contactine 1 et de CASPR1 sur l’axone et de neurofascine 155 sur la cellule gliale de la gaine de myéline consolide le contact entre les prolongements de myéline et l’axone (figure 1) [10].
Anti Contactin 1 (CNTN1) IgG
- Symptômes : 20 à 49% présentent un début subaigu de la maladie. Un début de symptôme agressif semblable au SGB est connu avec des parésies subaiguës (casuistique). Une neuropathie à prédominance motrice est typique et une ataxie sensitive est souvent observée. L’apparition de tremblements est plus fréquente que dans le cas d’une PIDC séronégative, mais moins fréquente que dans le cas d’une PIDC anti NF155 positive. L’âge de survenue de la maladie se situe autour de 50-60 ans. l’âge de 18 ans. Dans l’ensemble, il est décrit que les patients atteints de PIDC anti-CNTN1 positifs sont plus âgés au début de la maladie que les patients atteints de PIDC séronégatifs [2].
- Résultats : Les examens électroneurographiques révèlent des modifications axonales précoces au cours de la maladie (tableau 1).
- Implications thérapeutiques : Les patients ne répondent pas suffisamment aux immunoglobulines intraveineuses (IVIg). Les corticostéroïdes ont un bon effet. En cas de réponse insuffisante au traitement standard, un traitement de déplétion des cellules B par rituximab est efficace [14]. Il convient de noter qu’il est important que la durée de la maladie soit la plus courte possible afin de minimiser les dommages axonaux [8]. Sous immunothérapie efficace, les titres sériques d’anti CNTN1 diminuent [11].
Protéine 1 associée à l’anti-Contactine (CASPR1) IgG
- Symptômes : Des douleurs neuropathiques prononcées ont été décrites chez les patients séropositifs aux anti-CASPR1. Les symptômes commencent généralement de manière subaiguë avec une sévérité élevée et sont initialement accentués par la motricité. Anti CASPR1 a été détecté chez des patients atteints de CIDP et de SGB. Le début de la maladie est indiqué dans une description de cas vers l’âge de 30 ans.
- Résultats : Les examens électrophysiologiques mettent en évidence des blocs de conduction réversibles [11].
- Implications thérapeutiques : Une absence de réponse à l’IVIg et à la méthylprednisolone a été décrite. Le rituximab a montré une très bonne efficacité. Les douleurs neuropathiques associées s’améliorent avec un traitement efficace [2].
Anti Neurofascine 155 (NF155) IgG
- Symptômes : typiquement, un pourcentage élevé de patients CIDP NF155-positifs présentent une faiblesse accentuée au niveau distal. Les autres symptômes peuvent être un tremblement de haute amplitude et de basse fréquence et une ataxie sensitive avec des signes cérébelleux. L’association avec HLA-DRB1*15 est décrite [12]. L’âge de survenue de la maladie est nettement plus jeune que pour la CIPD séronégative, avec un âge de 20 à 30 ans au début de la maladie.
- Résultats : L’examen électrophysiologique révèle un schéma démyélinisant avec un allongement significatif des latences distales et des latences des ondes F (tableau 1). Une protéine fortement élevée a été détectée dans le LCR [13].
- Les implications thérapeutiques : La réponse aux IgIV est mauvaise. Une amélioration partielle a été décrite sous corticostéroïdes. Un bon succès thérapeutique est attendu sous rituximab et plasmaphérèse [14]. Sous une immunothérapie efficace, les titres sériques d’anti NF155 diminuent et une corrélation avec l’amélioration clinique a été démontrée [11].
Anti Neurofascine 140/186 (NF 140/186) IgG
- Symptômes : Les patients se présentent avec une polyradiculopathie sensitivomotrice symétrique d’évolution sévère. De plus, une ataxie sensitive et une atteinte des nerfs crâniens peuvent se produire. Certains patients présentaient une maladie auto-immune concomitante. L’âge de survenue de la maladie se situe autour de 50-60 ans. La plupart des cas ont été décrits avant l’âge de 40 ans [2].
- Résultats : Les électroneurographies ont mis en évidence des résultats démyélinisants avec des blocs de conduction (chez 3/5 patients) et des caractéristiques axonales (chez 2/5 patients) [15].
- Implications thérapeutiques : Sous IVIg et corticostéroïdes, les symptômes se sont partiellement améliorés. Une réponse potentiellement bonne a été décrite avec le rituximab [14].
Auto-anticorps dans les polyneuropathies inflammatoires aiguës – Syndrome de Guillain-Barré (GBS) avec les sous-types AIDP, AMAN et AMSAN
Localisation : Les canaux ioniques de l’anneau de Ranvier sont stabilisés par des gangliosides, des glycosphingolipides contenus dans la membrane cellulaire des régions nodale et paranodale [16]. Les différents sous-types du syndrome de Guillain-Barré sont chacun associés à des auto-anticorps dirigés contre différents gangliosides et se distinguent par leurs résultats électrophysiologiques.
La poly-neuropathie inflammatoire aiguë démyélinisante (PIAD) est le sous-type le plus fréquent du SGB en Europe et en Amérique du Nord. La plupart du temps, aucun auto-anticorps spécifique n’est détecté.
- Symptômes : La maladie est souvent précédée d’une infection gastro-intestinale ou d’une infection respiratoire. Les premiers symptômes sont des hypoesthésies, des paresthésies, des parésies et des douleurs aux extrémités. Les parésies sont bilatérales, symétriques et progressives. Les symptômes moteurs évoluent en 12 heures à 28 jours jusqu’à la tétraparésie avec implication des muscles respiratoires. De plus, des déficiences des nerfs crâniens et un dysfonctionnement autonome peuvent survenir [4].
- Résultats : ce sous-type démyélinisant de polyneuropathie inflammatoire aiguë est caractérisé par une réduction significative de la vitesse de conduction nerveuse, une augmentation des latences motrices distales, des blocs de conduction, une dispersion temporale anormale, des latences d’ondes F prolongées ou l’absence d’ondes F [17].
Anti LM1 (sialosylneolactotetraosylceramide)
Les auto-anticorps anti-LM1 (sialosylneolactotetraosylceramide) provoquent une polyneuropathie inflammatoire démyélinisante aiguë (PIDA) lorsqu’ils sont monospécifiques. Des cas ont été décrits dans lesquels les anticorps IgG anti-LM1 provoquent une réaction croisée avec des gangliosides tels que GM1, GalNAc-GD1a, GD1b et GQ1b, ce qui permet de poser le diagnostic d’AMAN ou d’AMSAN.
Anti GalC (Galactocérébroside)
Des autoanticorps anti-galactocérébroside (GalC) ont été détectés chez des enfants atteints de polyneuropathie inflammatoire démyélinisante aiguë sévère (PIDA) associée à la M. pneumoniae [18].
Anti Neurofascine 186, Anti Gliomedine, Anti NrCAM, Anti CNTN1, Anti Neurofascine 155, Anti CASPR1 et Anti CASPR2
Des études ont montré la présence d’auto-anticorps nodaux anti-Neurofascine 186, anti-Gliomédine et anti-N NrCAM, ainsi que d’auto-anticorps paranodaux anti-CNTN1, anti-Neurofascine 155 et anti-CASPR1 (voir la section CIDP ci-dessus) chez des patients adultes atteints de SGB. Des auto-anticorps juxtaparanodaux contre CASPR2 ont été décrits dans 2 cas de SGB chez des enfants.
La neuropathie axonale motrice aiguë (AMAN) est principalement présente en Asie [19].
- Symptômes : conformes à l’AIDP (voir ci-dessus) sans troubles de la sensibilité.
- Résultats : ce sous-type axonal de polyneuropathie inflammatoire aiguë se caractérise par une réduction significative de l’amplitude des potentiels d’action motrice cumulés (CMAP) et par ce que l’on appelle un “échec réversible de la conduction”, c’est-à-dire qu’une amplitude CMAP réduite et des blocs de conduction peuvent se rétablir soudainement lors d’examens électroneurographiques répétés, sans qu’une dispersion temporelle ne soit détectable comme signe de remyélinisation. [16].
Anti-GM1 IgG, Anti-GM2, Anti-GD1b IgG, Anti-GT1b, Anti- GM3, Anti-GD1a IgG et Anti-GalNac-GD1a
Les anticorps anti-gangliosides anti-GM1 IgG, anti-GM2, anti-GD1b, anti-GT1b, anti-GM3, anti-GD1a IgG et anti-GalNac-GD1a peuvent être détectés chez environ 80% des patients atteints d’AMAN. Ces auto-anticorps peuvent être isolés ou combinés.
La neuropathie axonale aiguë motrice et sensitive (AMSAN)
- Symptômes : voir AIDP
- Résultats : mêmes critères que pour l’AMAN plus réduction de l’amplitude du potentiel d’action nerveux sensitif (Unicini et al. 2018).
anti-GM1 IgG, anti-GM1b, anti-GD1a IgG
Les anticorps anti-gangliosides anti-GM1, anti-GM1b, anti-GD1a sont détectables dans la neuropathie axonale aiguë motrice et sensitive (ASMAN) [19].
Auto-anticorps dans le syndrome de Miller-Fisher (MFS)
Localisation : Le ganglioside GQ1b se trouve principalement dans la myéline paranodale des nerfs crâniens qui innervent les muscles -oculaires [19].
Anti GQ1b IgG
- Symptômes : L’ophtalmoplégie aiguë, la neuropathie ataxique aiguë et l’aréflexie sont les principaux symptômes du syndrome de Miller-Fisher. Dans le cas de l’encéphalite du tronc cérébral de Bickerstaff, il existe, en plus de l’ophtalmoplégie et de l’ataxie, un trouble de la conscience et généralement une hyperréflexie.
Des auto-anticorps GQ1b peuvent être détectés chez 90% des patients atteints du syndrome de Miller-Fisher (MFS). Le syndrome de Miller-Fisher et l’encéphalite du tronc cérébral de Bickerstaff sont associés aux anti-GQ1b, de sorte que Shahrizaila et Yuki ont regroupé ces maladies sous le nom de syndrome des anticorps anti-GQ1b [20].
Auto-anticorps dans la neuropathie motrice multifocale (MMN)
Localisation : le ganglioside GM1 est principalement localisé dans la région nodale de l’anneau de Ranvier à lacet. Les auto-anticorps se lient dans la région nodale et activent le complément, ce qui affecte le clustering des canaux sodiques.
Complexe anti-GM1 IgM et anti-Galactocérébroside GM1
- Symptômes : les caractéristiques sont des parésies des extrémités, généralement asymétriques, qui augmentent lentement et commencent souvent aux extrémités supérieures. On ne trouve pas de déficits sensibles. De plus, un tremblement de maintien, des fasciculations et des crampes peuvent apparaître.
- Résultats : des anticorps IgM anti-GM1 sont détectables chez 50% des patients atteints de MMN [21]. Les examens électrophysiologiques permettent de mettre en évidence des blocs de conduction. L’activation du complément peut entraîner une lésion axonale.
- Implications thérapeutiques : Bonne réponse aux IgIV. La détection d’auto-anticorps peut étayer le diagnostic de MMN si tous les critères ne sont pas remplis et aider à mettre en place un traitement efficace par IgIV. Cliniquement, il existe une maladie du 2e motoneurone, de sorte que la MMN constitue un diagnostic différentiel important de la sclérose latérale amyotrophique [11].
Auto-anticorps dans la polyneuropathie MGUS (MGUS-P)
Localisation : la glucoprotéine associée à la myéline (MAG) est localisée dans la myéline de la région paranodale.
Anti MAG (glucoprotéine associée à la myéline) IgM
- Symptômes : une polyneuropathie distale à progression lente, principalement sensitive et ataxique, est caractéristique. Les extrémités distales supérieures sont souvent touchées [11]. La plupart des patients ont moins de 50 ans.
- Résultats : L’examen électrophysiologique révèle un schéma démyélinisant. L’immunofixation révèle une gammapathie monoclonale IgM. Chez 50% des patients atteints de MGUS-P, les anticorps anti-MAG IgM sont positifs. Le niveau du titre d’anticorps ne semble pas être corrélé à la sévérité de la maladie et à la réponse au traitement. La détection des anticorps anti-MAG n’est nécessaire que pour établir le diagnostic.
- Implications thérapeutiques : Quelques études ont montré une réponse à la plasmaphérèse, au cyclophosphamide, à l’IVIg et au rituximab. Il existe des preuves qu’une déplétion précoce des cellules B avec le rituximab peut influencer la progression [11].
Auto-anticorps associés à des maladies immunitaires systémiques
Anticorps SSA (Ro) et SSB (La), anticorps antineuronaux, anticorps FGFR3 (fibroblast growth factor receptor 3)
- Symptômes : Chez les (jeunes) patients présentant une neuropathie/neuronopathie sensitive progressive, aiguë ou subaiguë, il est utile de rechercher un syndrome de Sjögren ou de dépister une maladie auto-immune [22].
- Résultats des examens : Le LCR montre une légère augmentation de la protéine avec un nombre normal de cellules. Des anomalies sont détectables électrophysiologiquement dans les nerfs sensitifs qui ne sont pas dépendants de la longueur. Une perte étendue ou de faibles amplitudes de potentiels d’action nerveux sensibles est typique, de même qu’une distribution clinique asymétrique (“patchy”). Occasionnellement, les nerfs sensitifs des bras peuvent être plus affectés que ceux des jambes [23].
- Les implications thérapeutiques : La maladie sous-jacente doit être traitée.

Devant quelle présentation clinique faut-il penser à une genèse médiée par des auto-anticorps ?
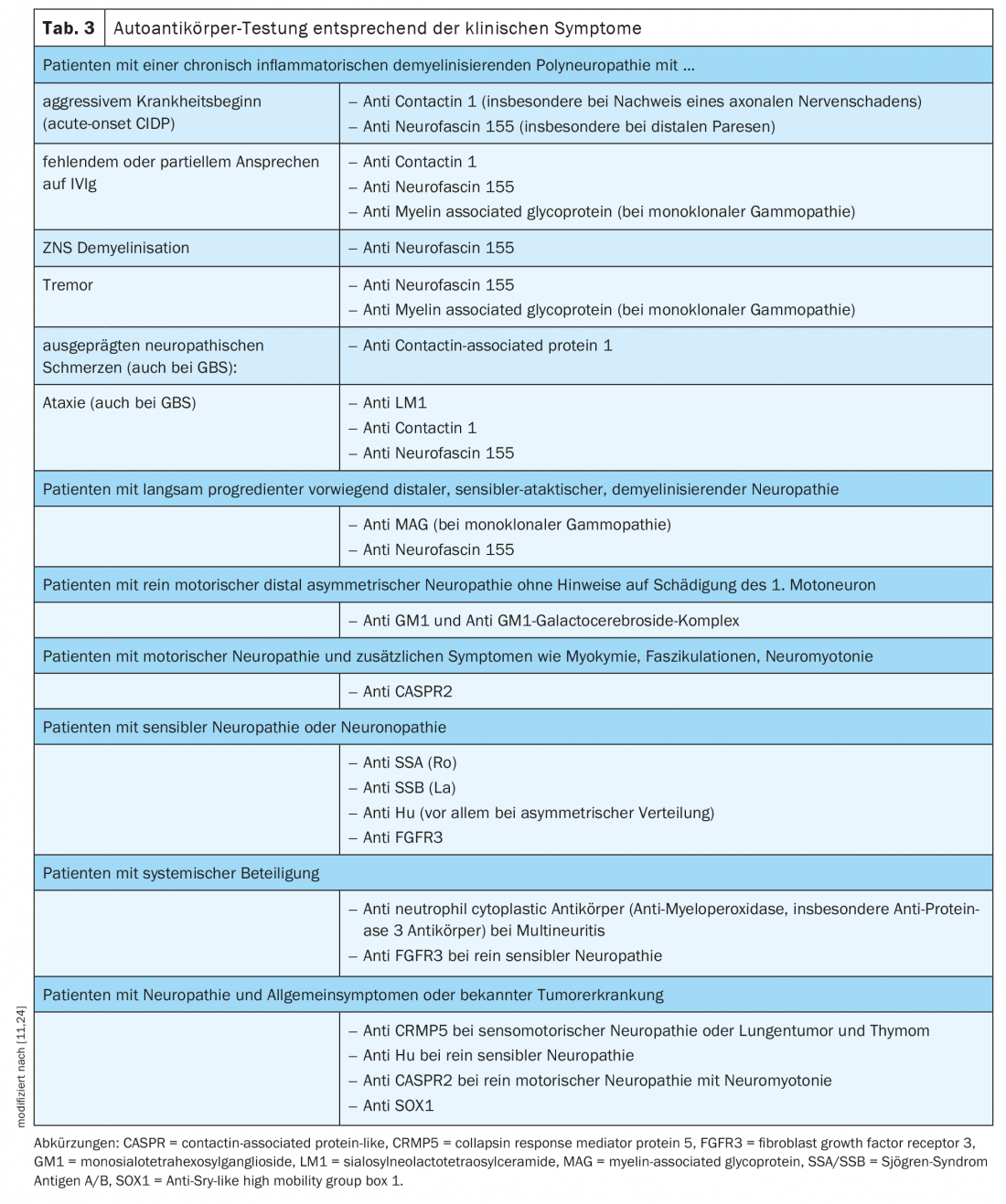
La recherche d’auto-anticorps nodaux et paranodaux doit être effectuée chez les patients présentant une polyneuropathie démyélinisante acquise aiguë, subaiguë ou chronique, associée à des symptômes supplémentaires tels que des tremblements, une atteinte distale ou une mauvaise réponse aux IgIV [11] (tableau 3).
Pourquoi le traitement standard est-il souvent inefficace dans la polyneuropathie inflammatoire démyélinisante chronique séropositive ?
Des études ont montré que les auto-anticorps nodaux et paranodaux appartiennent à différentes sous-classes d’IgG (tableau 4). Les IgG2 et les IgG3 ont été trouvées dans les SGB monophasiques séropositifs aux anti-CNTN1 et CASPR1, tandis que les IgG4 n’ont été trouvées jusqu’à présent que dans les cas d’évolution chronique. Par Appeltshauser et al. il a été décrit qu’un changement de classe d’IgG3 à IgG4 peut se produire au cours de la maladie dans les neuropathies séropositives anti-CNTN1 et anti-CASPR1 [25]. Actuellement, les sous-classes d’IgG des auto-anticorps nodaux et paranodaux ne sont déterminées que dans les laboratoires de recherche.

Les neuropathies médiées par les IgG3 répondent bien au traitement standard par IVIg. L’IgIV agit sur les polyneuropathies inflammatoires, notamment en inhibant le complément et en neutralisant les anticorps. Les patients présentant des anticorps IgG2 et IgG4 ne présentent aucune amélioration sous perfusion d’IVIg ou la réponse diminue au cours de la maladie. La mauvaise réponse ou l’absence de réponse aux IVIg dans les maladies à médiation IgG4 est attribuée à la faible capacité à lier les récepteurs FcγIIb et à l’absence d’activation du complément [14]. Dans les maladies auto-immunes neurologiques et non neurologiques médiées par les IgG4, telles que la myasthénie grave anti-musculaire, la pancréatite auto-immune et la cholangite sclérosante, l’efficacité du rituximab a été démontrée dans de nombreuses études. L’effet de déplétion des cellules B du rituximab est crucial dans les maladies à médiation IgG4. Une réponse partielle aux corticostéroïdes a été rapportée chez les patients présentant des anticorps IgG4 nodaux et paranodaux [14]. Les neuropathies positives aux auto-anticorps nécessitent un traitement rapide et efficace avant que des dommages irréversibles ne surviennent. En cas d’absence de réponse au traitement standard (IVIg & stéroïdes) pour une polyneuropathie inflammatoire démyélinisante chronique, un traitement par rituximab doit être envisagé.
Messages Take-Home
- Parmi les neuropathies à médiation immunitaire, le diagnostic différentiel doit être posé avec les nodo-/paranodopathies, car il existe des options thérapeutiques efficaces.
- La chimie de laboratoire permet de détecter les auto-anticorps dirigés contre les protéines nodales et paranodales de l’anneau de Ranvier dans les nodo- et paranodopathies.
- Les caractéristiques électroneurographiques sont définies pour les nodo- et paranodopathies.
- Le diagnostic des neuropathies associées à des auto-anticorps repose sur des examens électroneurographiques (modèle démyélinisant ou axonal, caractéristiques des nodopathies et des paranodopathies) et sur des diagnostics de laboratoire et du LCR (exclusion des diagnostics différentiels pertinents, détection d’une dissociation cytalbumine dans le LCR) ; l’échographie nerveuse (modèle de gonflement des nerfs) est en outre utile.
- Tester les auto-anticorps nodaux et paranodaux à partir du sérum (pas de production intrathécale et titres faibles dans le LCR).
- En l’absence d’amélioration sous traitement de première ligne (IVIg, stéroïdes) et chez les patients présentant une évolution aiguë ou subaiguë d’une polyneuropathie démyélinisante acquise avec des symptômes supplémentaires tels que tremblements, ataxie et atteinte distale (voir tableau 2 : Red Flags in CIDP), envisager la détermination des auto-anticorps et, si nécessaire, un traitement par rituximab.
- Sélection des auto-anticorps sur la base des symptômes cliniques (tableau 3).
Littérature :
- Visser NA : Incidence de la polyneuropathie à Utrecht, aux Pays-Bas. Neurology 2015, Jan 20 ; 84(3) : 259-264.
- Vural A, Doppler K : Autoantibodies against the node of Ranvier in seropositive chronic inflammatory demyelinating polyneuropathy : diagnostic, pathogenic, and therapeutic relevance. Front. Immunol. 2018, May 14 ; 9 : 1029
- Stathopoulos P : Cibles antigéniques auto-immunes au niveau du ganglion de Ranvier dans les troubles démyélinisants. Nat Rev Neurol. 2015 Mar ; 11(3) : 143-156.
- Kieseier BC, Mathey EK, Sommer C : Neuropathies à médiation immunitaire. Nat Rev Dis Primers 2018 4, 31.
- Svennerholm L : Ganglioside designation. Adv Exp Med Biol. 1980 ; 125 : 11
- Grether NB, et al : Diagnostic des polyneuropathies à médiation immunitaire. DGNeurologie 2020 ; 3 (2) : 147-158.
- Uncini A, Kuwabara S : Critères d’électrodiagnostic pour le syndrome de Guillain-Barre : une révision critique et la nécessité d’une mise à jour. Clin Neurophysiol, 2012, 123(8), 1487-1495.
- Uncini A, Kuwabara S : Nodopathies du nerf périphérique : un concept émergent. J Neurol Neurosurg Psychiatry, 2015, 86(11), 1186-1195.
- Uncini A, Susuki K, Yuki N : Nodo-paranodopathy : au-delà de la classification démyélinisante et axonale dans les neuropathies médiées par les anticorps anti-gangliosides. Clin Neurophysiol, 2013, 124(10), 1928-1934.
- Poliak S, Peles E : La différenciation locale des axones myélinisés aux nœuds de Ranvier. Nat Rev Neurosci, 2003, 4(12), 968-980.
- Querol L : Autoantibodies in chronic inflammatory neuropathies : diagnostic and therapeutic implications, Nat Rev Neurol. Neurology, 2017, Sep ; 13(9) : 533-547.
- Martinez-Martinez L : Anti-NF155 chronic inflammatory demyelinating polyradiculoneuropathy strongly associates to HLA-DRB15. J Neuroinflammation 2017 ; 14 : 224.
- Kadoya M : IgG4 anti-neurofascine155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy : Clinical significance and diagnostic utility of a conventional assay. Journal of Neuroimmunology 2016 Dec 15 ; 301 : 16-22.
- Bunschoten C : Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy, Lancet Neurology 2019 ; 18 : 784-94.
- Delmont E, Manso C, Querol L, et al : Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Cerveau . 2017 Jul 1;140(7) : 1851-1858.
- Susuki K : Les gangliosides contribuent à la stabilité des jonctions paranodales et des clusters de canaux ioniques dans les fibres nerveuses myélinisées. Glia, 2007a, 55(7) : 746-757.
- Uncini A, Kuwabara S : L’électrodiagnostic des sous-types du syndrome de Guillain-Barré : Où en sommes-nous ? Neurophysiologie clinique. 2018 Dec ; 129(12) : 2586-2593.
- Meyer Sauteur PM : Mycoplasma pneumoniae déclenchant le syndrome de Guillain-Barré : une étude cas-témoins, Ann Neurol 2016 Oct ; 80(4) : 566-580.
- Pei S : Variantes axonales du syndrome de Guillain-Barré : une mise à jour. Springer Nature 2020 March.
- Shahrizaila N, Yuki N : Bickerstaff brainstem encephalitis and Fisher syndrome : anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013, 84 : 576-583.
- Van Asseldonk JT : Multifocal motor neuropathy. Lancet Neurol, 2005, 4(5) : 309-319.
- Heuss D : Diagnostic des polyneuropathies, ligne directrice S1, 2019, dans : Société allemande de neurologie (éd.), Lignes directrices pour le diagnostic et le traitement en neurologie. En ligne : www.dgn.org/leitlinien. (consulté le 01.04.2021).
- Sghirlanzoni A : Maladies des neurones sensoriels. The Lancet Neurology. 2005 ; 4(6) : 349-361
- Sun X : Anti-SOX1 Antibodies in Paraneoplastic Neurological Syndrome. J Clin Neurol. 2020 Oct;16(4):530-546.
- Appeltshauser L : Antiparanodal antibodies and IgG subclasses in acute autoimmune neuropathy. Neurol Neuroimmunol Neuroinflamm. 2020 Juillet 24 ; 7(5) : e 817.
- Ilyas A : Immunoglobuline G subclass distribution of autoantibodies to gangliosides in patients with Giullain-Barre syndrome. Res Commun Mol Pathol Pharmacol. 2001 juillet ; 109(1-2) : 115-123.
- Lardone RD : Neurological disorders-associated anti-glycosphingolipid IgG-antibodies display differentially restricted IgG subclass distribution. Sci Rep. 2020 Aug 4;10(1) : 13074.
InFo NEUROLOGIE & PSYCHIATRIE 2021 ; 19(3) : 19-25