Les symptômes non spécifiques au stade initial entraînent une nette latence diagnostique de ces lymphomes cutanés primaires à cellules T. Les symptômes peuvent être très variables d’une personne à l’autre. La photophérèse extracorporelle et les nouvelles thérapies ciblées améliorent de plus en plus le succès thérapeutique du syndrome de Sézary.
Le syndrome de Sézary est une maladie agressive rare, souvent difficile à diagnostiquer, appartenant au groupe des lymphomes cutanés primaires à cellules T (CTCL) [1,2]. Cette maladie fait partie des lymphomes non hodgkiniens extranodaux et se caractérise par la triade érythrodermie prurigineuse, lymphadénopathie généralisée et cellules T néoplasiques à noyaux cérébriformes (cellules de Sézary) dans le sang, la peau et les ganglions lymphatiques [3,25]. Le syndrome de Sézary, dont l’incidence est de 0,1/100 000, survient principalement entre 40 et 60 ans . Il s’agit d’une maladie rare qui se caractérise par l’apparition d’une tumeur au cerveau. Les hommes et les Afro-Américains sont plus souvent touchés que les femmes et les Caucasiens [4,5].
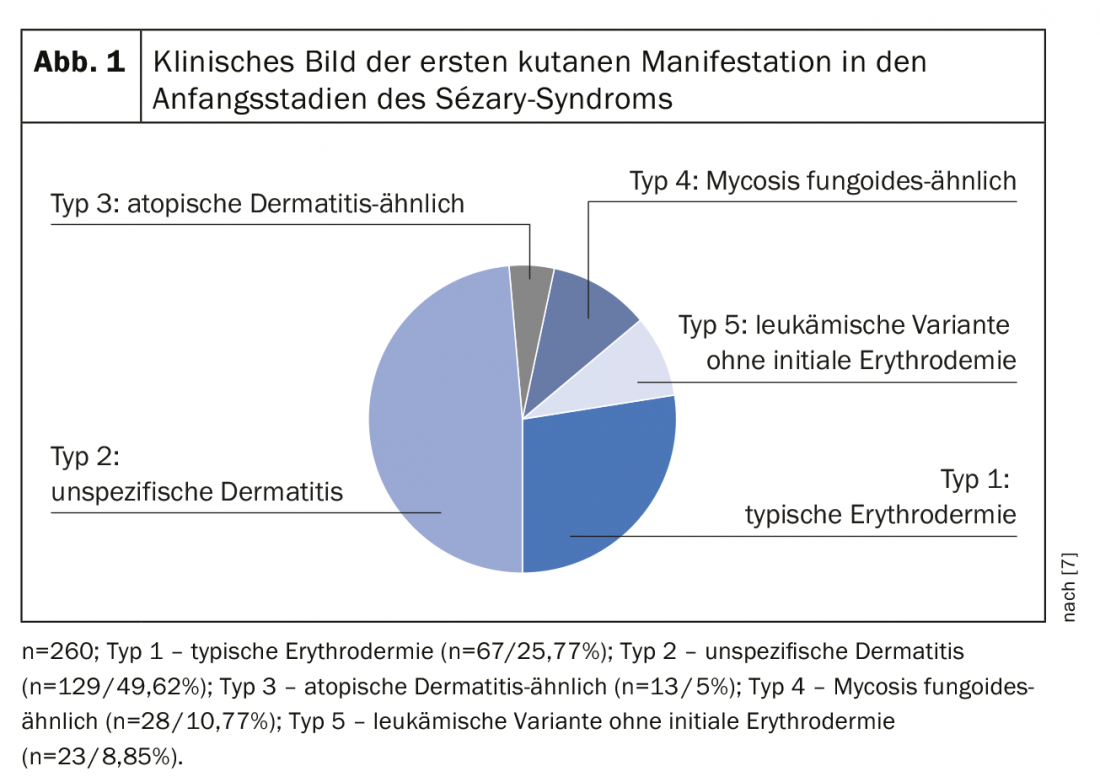
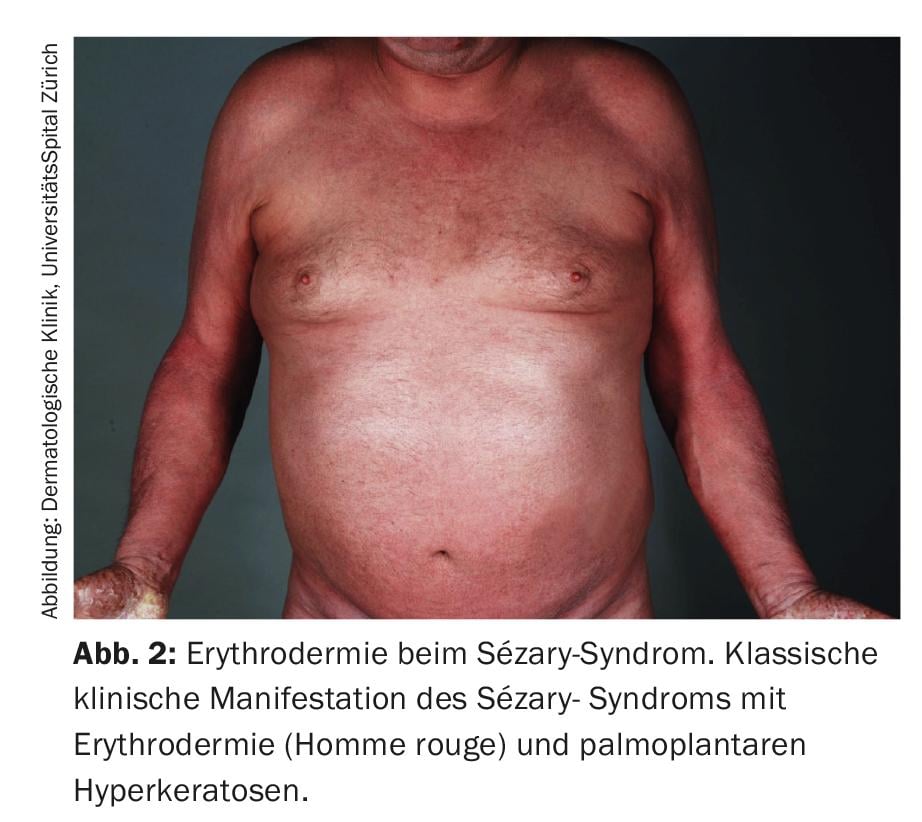
La définition du syndrome de Sézary par la Société internationale des lymphomes cutanés en collaboration avec l’Organisation mondiale de la santé inclut l’érythrodermie comme symptôme obligatoire accompagné d’au moins deux des manifestations suivantes : >1000 cellules de Sézary/µL dans le sang périphérique, anomalies immunophénotypiques des cellules T Ratio CD4/CD8 ≥10, cellules CD4+/CD7- ≥30%, ou cellules CD4+/CD26- ≥ 40 [25]La présence d’une population de récepteurs de cellules T monoclonales (TZR) dans le sang ou de clones de cellules T chromosomiquement modifiés [6]. Une étude de cohorte rétrospective publiée par Mangold et al. a toutefois montré que seuls 25,5% des patients présentaient une érythrodermie typique, qui touche >80% de la surface corporelle, comme première manifestation. Cependant, chez 86,3% des patients, l’érythrodermie se développe au fil du temps et est présente au plus tard au moment du diagnostic. La présentation clinique initiale, souvent non spécifique, est donc responsable d’un retard diagnostique allant d’un mois à 32 ans (moyenne de 4,2 ans) (Fig. 1) [7]. Parmi les autres symptômes du syndrome de Sézary figurent l’alopécie, l’onychodystrophie, l’hyperkératose palmoplantaire ainsi que des démangeaisons massives, qui sont une source de grande souffrance pour les patients concernés (fig. 2). La perte de l’intégrité cutanée s’accompagne d’un risque accru d’infection par la flore cutanée résidente, comme le staphylocoque doré. Les infiltrations cutanées tumorales, associées à des œdèmes ainsi qu’à une hypalbuminémie, peuvent entraîner une déshydratation.
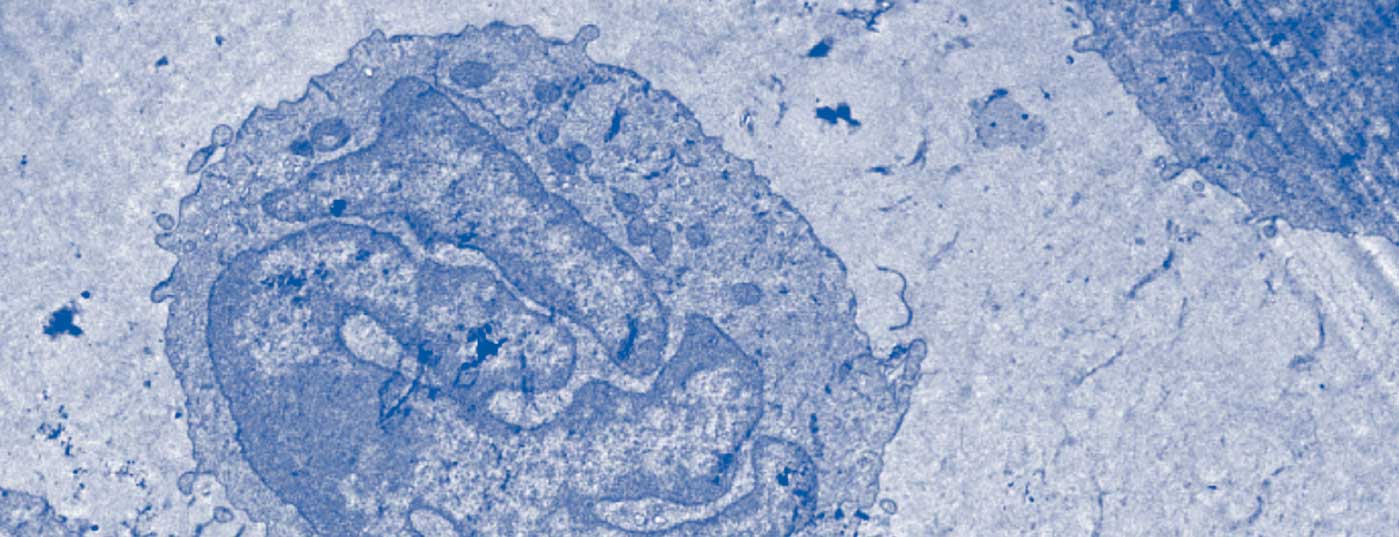
Diagnostic
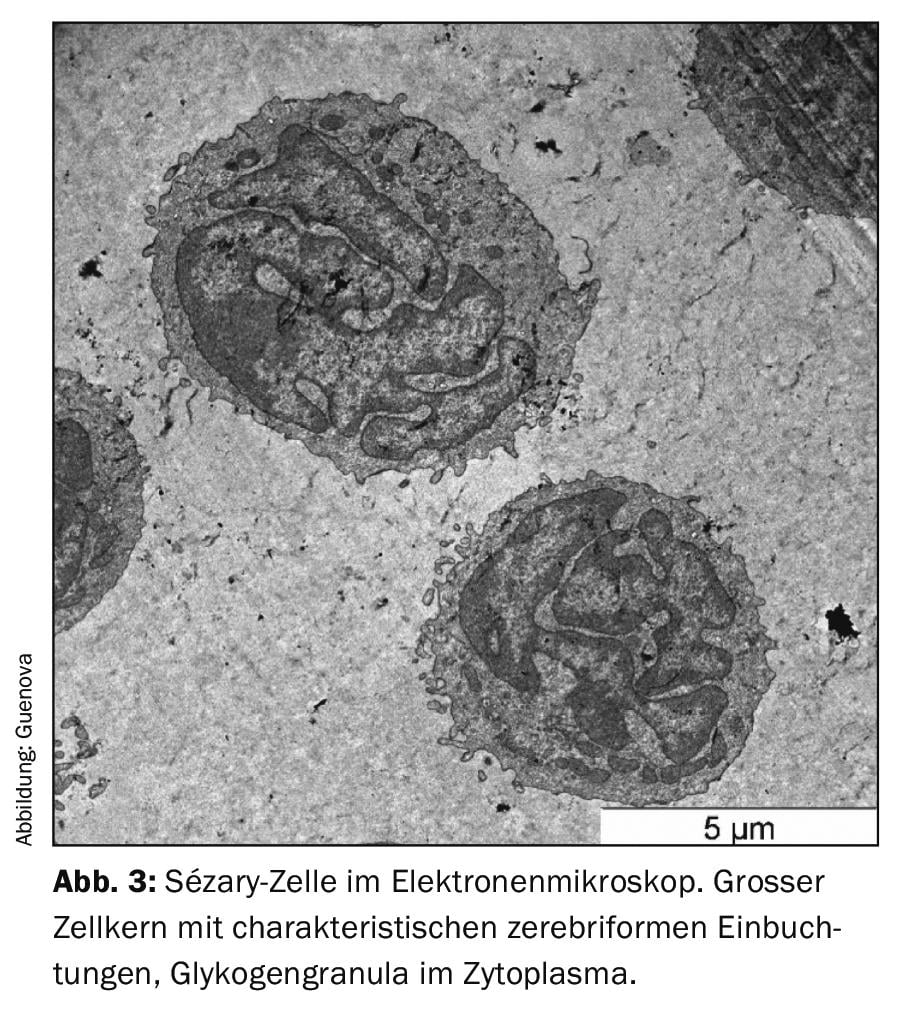
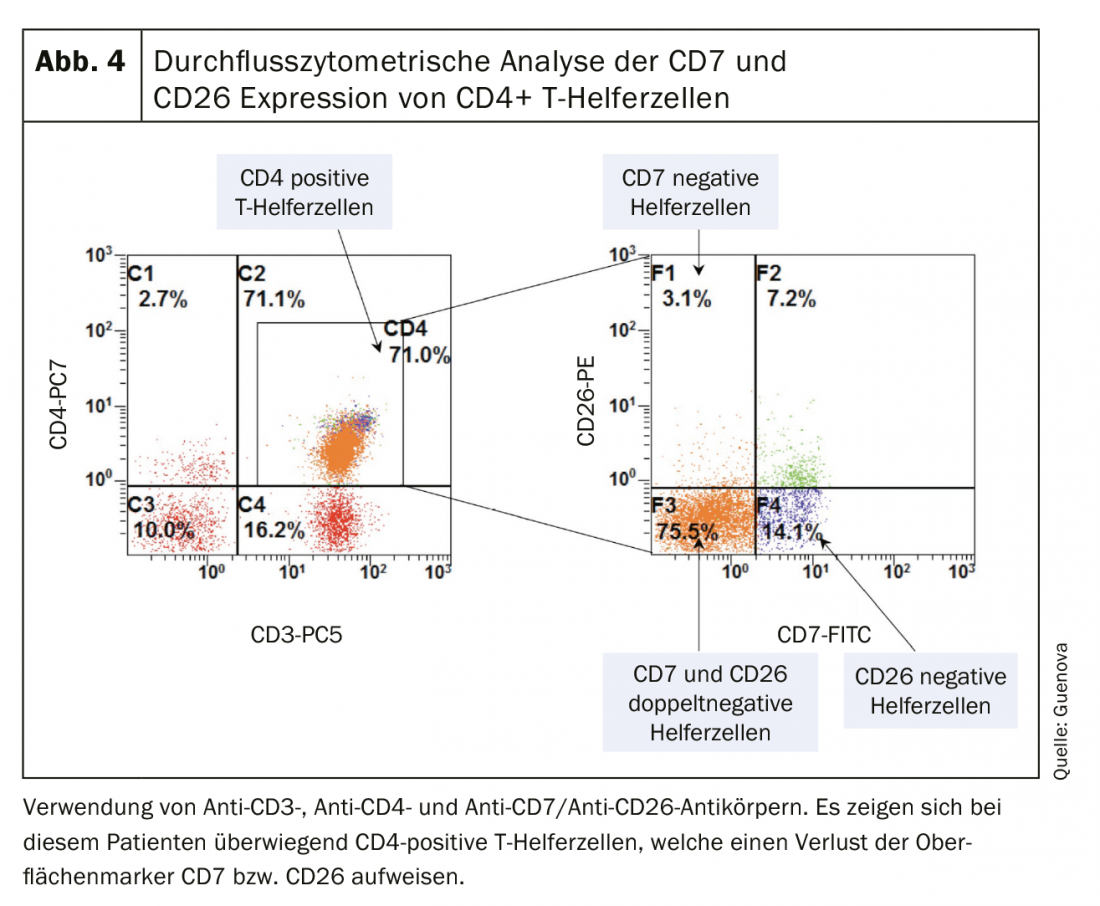
Comme pour de nombreuses maladies érythrodermiques, l’absence de marqueurs diagnostiques clairs rend le diagnostic différentiel du syndrome de Sézary difficile [3,8]. L’examen clinique indispensable comprend, outre un relevé très précis de l’état cutané, une palpation de toutes les régions ganglionnaires. L’outil d’évaluation pondérée de la peau modifiée (Modified Skin Weighted Assessment Tool, mSWAT) est particulièrement adapté pour mesurer et évaluer l’atteinte cutanée [8,9]. Des biopsies multiples de lésions cutanées représentatives sont examinées par des méthodes histopathologiques et immunohistochimiques [3]. L’histologie révèle des infiltrats monomorphes de cellules T atypiques, une épidermotropie caractéristique et des micro-abcès de Pautrier – des amas intra-épidermiques de cellules malignes [8]. Cependant, dans environ 40% des cas, les résultats histologiques ne sont pas clairs pour le diagnostic [10]. Le sang périphérique est analysé par cytométrie de flux et par des techniques moléculaires ou cytogénétiques afin de détecter la présence d’une population anormale de lymphocytes T CD4+ et de lymphocytes T néoplasiques morphologiquement anormaux (frottis sanguin). Ces cellules de Sézary (ou cellules de Lutzner) typiques sont caractérisées par une morphologie ultrastructurale avec un noyau cérébriforme. (Fig.3). Ils expriment non seulement les récepteurs de chimiokines CCR7 et CCR4, mais présentent également une expression accrue de la L-sélectine et un déficit caractéristique des antigènes de surface CD7 et CD26. (ill.4) [3,11]. La mise en évidence d’une clonalité du récepteur des cellules T a une grande valeur diagnostique [12]. La norme est la détection moléculaire par réaction en chaîne par polymérase [13]. Les analyses par cytométrie de flux de l’antigène du récepteur des cellules T Vβ sont utilisées en routine dans les centres universitaires spécialisés [12].
Des analyses récentes ont montré une expression accrue de l’antigène de surface CD164 dans le nombre total de cellules T CD4+ chez les patients atteints du syndrome de Sézary. Ce marqueur pourrait donc devenir à l’avenir un paramètre diagnostique prometteur et une cible thérapeutique potentielle [14,15]. D’autres nouveaux biomarqueurs sanguins et cutanés, notamment PD-1 (CD279) et KIR3DL2 (CD158k), peuvent faciliter la distinction entre le syndrome de Sézary et les dermatoses inflammatoires érythrodermiques [25].
Staging et pronostic
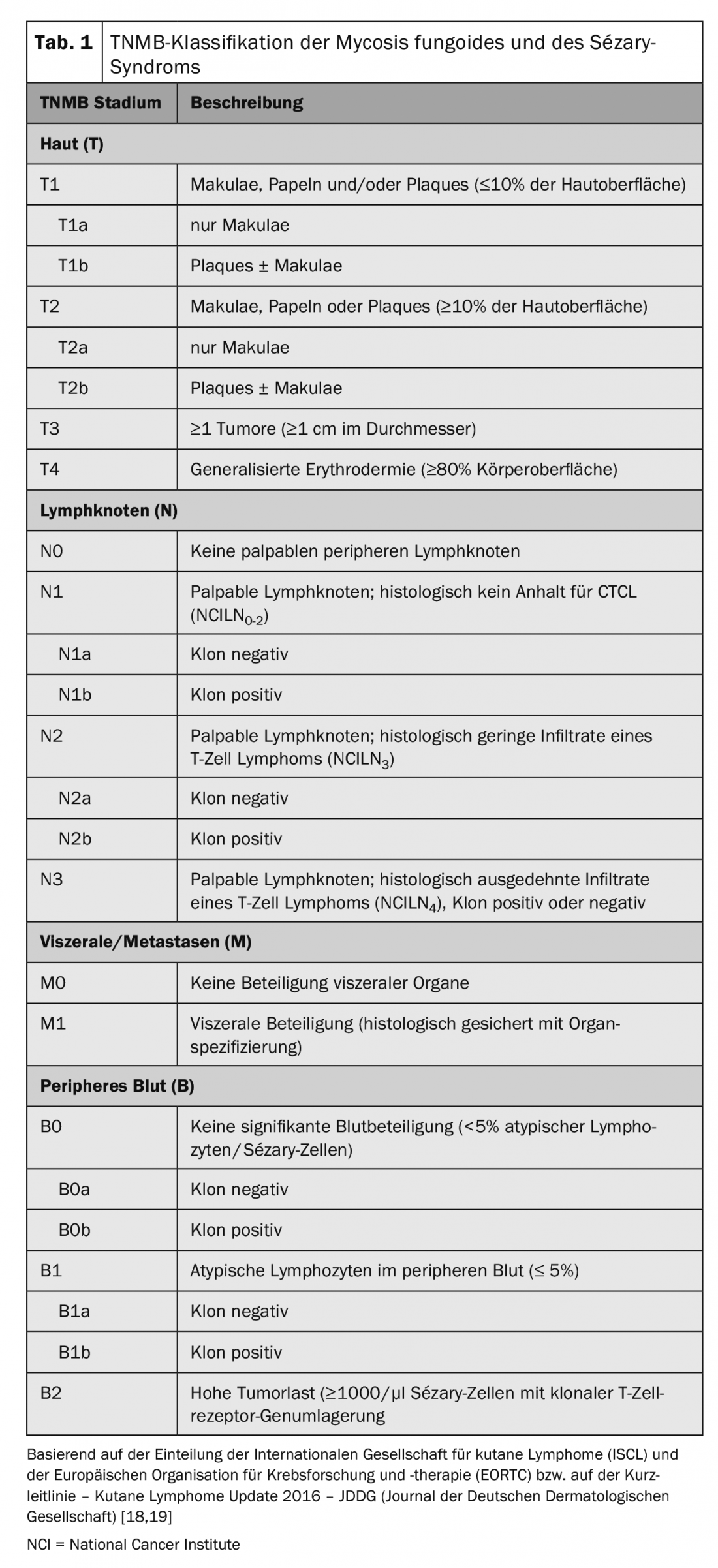
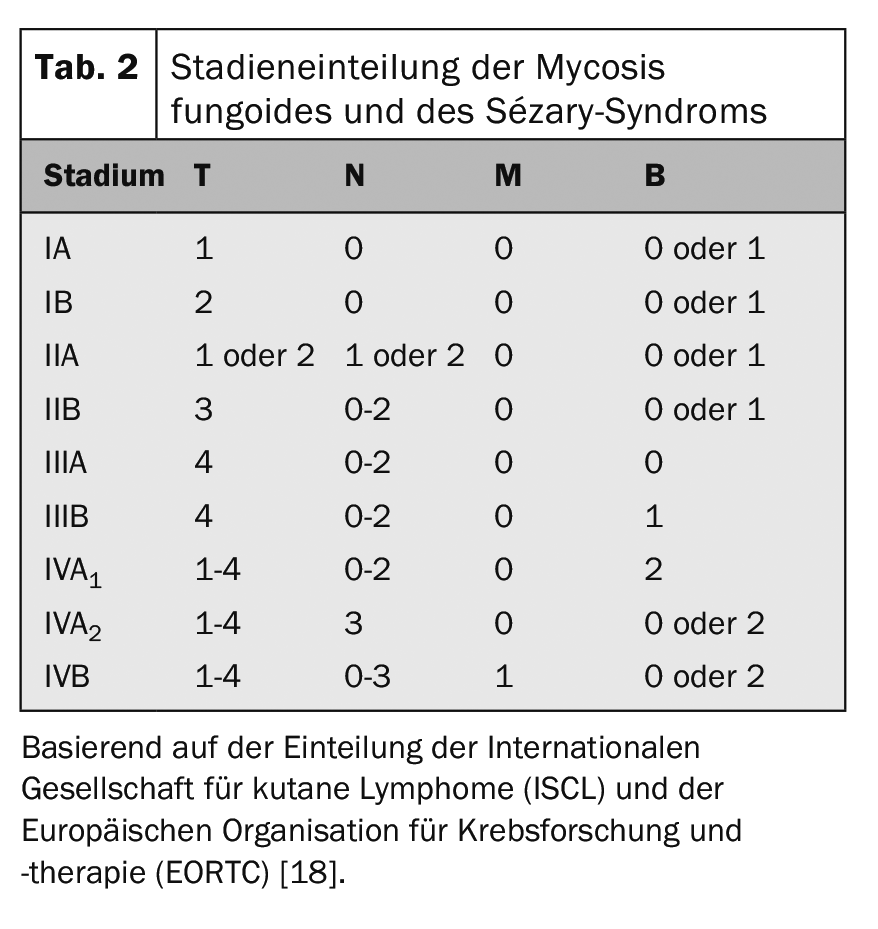
La classification TNM internationalement reconnue est utilisée pour le staging des tumeurs CTCL (tab. 1). Celle-ci comprend, outre l’atteinte cutanée – tumeur primaire (T), atteinte ganglionnaire (N) et métastases à distance (M) – la détection de cellules de Sézary dans le sang périphérique (B) et est donc appelée classification TNMB (tab. 1). Cette classification sert de base à la répartition des stades (tab. 2). Le syndrome de Sézary, avec son implication sanguine obligatoire, correspond donc toujours au stade IV [16]. En conséquence, la maladie présente un mauvais pronostic avec un taux de survie moyen de <3 ans. Outre la participation du sang périphérique, d’autres facteurs pronostiques négatifs ont été identifiés : des phénotypes atypiques ainsi que la perte caractéristique des caractéristiques de surface CD7 et CD26 [17].
Les examens d’imagerie tels que le scanner corps entier et l’échographie des ganglions lymphatiques dans le cadre du staging initial de la tumeur sont recommandés pour tous les patients à partir du stade T2 [8,20]. Un PET-CT ainsi qu’une biopsie des ganglions lymphatiques doivent être effectués chez les patients chez lesquels une lymphadénopathie et/ou une extension systémique sont suspectées [8,16]. Une biopsie de la moelle osseuse doit être effectuée chez les patients présentant des anomalies hématologiques. Un diagnostic élargi ainsi que des biopsies d’organes viscéraux peuvent être envisagés en cas de suspicion d’une atteinte extracutanée [8]. Les patients atteints du syndrome de Sézary doivent être pris en charge de manière optimale par une équipe multidisciplinaire composée de dermatologues, d’oncologues, de dermatopathologues et de radiologues, tant sur le plan diagnostique que thérapeutique [16].
Étiologie
Bien que la cause de cette maladie complexe ne soit pas encore totalement comprise à ce jour, on peut supposer que la physiopathologie du syndrome de Sézary s’explique d’une part par un dysfonctionnement immunitaire et d’autre part par des modifications épigénétiques [21]. Jusqu’à récemment, on pensait que le caryotype du syndrome de Sézary était caractérisé par des délétions concernant les chromosomes 10q et 17p et par des insertions des chromosomes 8q, 10p et 17q [8]. Une étude menée par Izykowska et al. montre que les altérations malignes se situent cependant à des niveaux génétiques très différents [21,22]. Comme dans la plupart des CTCL leucémiques, il existe dans le syndrome de Sézary une réponse immunitaire à prédominance Th2 [23]. Les cytokines de type Th2 sécrétées en grande quantité, l’interleukine 4 (IL-4), l’IL-5, l’IL-13, l’IL-21 et l’IL-31, suppriment la réponse immunitaire médiée par Th1 et servent également de cible pharmacologique [3,21].
Diagnostic différentiel
Le diagnostic différentiel entre le syndrome de Sézary et le mycosis fongoïde (MF), le plus fréquent de tous les CTCL, est particulièrement difficile à établir. Sur le plan clinique et diagnostique, les deux maladies présentent de nombreuses similitudes. Campbell et al. ont cependant pu montrer que le syndrome de Sézary et la MF proviennent de sous-groupes fonctionnels de lymphocytes T différents et qu’ils doivent donc être considérés comme des lymphomes distincts [3,24]. Alors que l’implication sanguine est inexistante ou minime dans la MF, elle représente un élément essentiel dans le syndrome de Sézary [7]. Parmi les autres diagnostics différentiels non néoplasiques du syndrome de Sézary figurent l’érythrodermie psoriasique, la dermatite atopique ou d’autres formes de dermatite, le pytiriasis rubra pilaire, les réactions médicamenteuses et l’érythrodermie idiopathique. La distinction entre le syndrome de Sézary au stade précoce et les dermatoses inflammatoires érythrodermiques peut constituer un défi.
Thérapie
Le traitement du syndrome de Sézary, qui ne permet toujours pas d’obtenir une guérison complète, dépend principalement de l’étendue de la maladie, de son impact sur la qualité de vie, des facteurs pronostiques ainsi que de l’âge du patient et des comorbidités [16]. Selon la dernière mise à jour du JDDG sur le traitement du syndrome de Sézary, la photophérèse extracorporelle (PCE), qui présente peu d’effets secondaires, fait partie des mesures thérapeutiques de premier choix. La PEC peut être utilisée en monothérapie ou en combinaison avec des corticostéroïdes topiques, la photothérapie par psoralènes avec UV-A (PUVA), ou par voie systémique avec l’interféron alpha (INF-α) ou le bexarotène [19,26].
Comme traitement de seconde ligne, on recommande de faibles doses de méthotrexate, un antagoniste de l’acide folique, des rétinoïdes administrés par voie systémique (bexarotène ; tous deux également combinés à la PUVA et à l’ECP), le chlorambucil, un cytostatique, en combinaison avec un glucocorticoïde à faible dose, ainsi que l’irradiation électronique de la peau entière [19,20,26]. Dans les stades avancés du syndrome de Sézary, la monochimiothérapie par gemcitabine ou doxorubicine liposomale pégylée fait partie des traitements de deuxième intention.
Ces dernières années, le succès thérapeutique s’est progressivement amélioré grâce aux thérapies ciblées contre le cancer (“targeted therapy”). L’alemtuzumab, un anticorps monoclonal humanisé anti-CD52, a été évalué dans une étude de phase II sur 22 patients atteints de MF/SS avancée, avec des taux de réponse de 55% [27].
En raison du risque significativement accru de complications infectieuses, une administration sous-cutanée du médicament à faible dose a été étudiée dans une cohorte de 14 patients atteints de SS réfractaire, avec des taux de réponse supérieurs à 80% [28]. Le conjugué anticorps-médicament brentuximab vedotin n’est pas encore autorisé pour le syndrome de Sézary, mais il peut éventuellement être utilisé hors indication dans les variantes CD30+ du syndrome de Sézary. Le pralatrexate, un antagoniste de l’acide folique, est autorisé pour le lymphome périphérique à cellules T. Il s’agit d’un traitement de première intention pour les patients atteints de lymphome à cellules T. Une étude clinique randomisée de phase III comparant le mogamulizumab, un anticorps monoclonal CCR4, au vorinostat (MAVORIC) a montré une amélioration significative de la survie sans progression chez les patients MF / SS randomisés pour recevoir le mogamulizumab. Chez les patients atteints du syndrome de Sézary, la réponse générale était la plus élevée (37%) [29].
Plusieurs inhibiteurs de l’histone déacétylase (HDAC), qui interviennent dans la régulation épigénétique de la transcription, sont déjà autorisés hors d’Europe pour le traitement du syndrome de Sézary [20]. En Europe, le resminostat, un inhibiteur de l’HDAC, est actuellement testé dans le cadre d’essais cliniques contre placebo en tant que traitement d’entretien pour les patients atteints de SS et MF avancés. Les inhibiteurs de points de contrôle immunitaires anti-PD1 et anti-CTLA4 sont aujourd’hui approuvés pour le traitement des tumeurs solides multiples, y compris le mélanome. Les données préliminaires d’une étude de phase II sur le pembrolizumab (anticorps monoclonal anti-PD1) dans la MF et la SS récidivantes/réfractaires semblent prometteuses avec un taux de réponse de 33%, y compris chez les patients présentant une maladie avancée [30].
La seule option de traitement ayant un potentiel curatif est actuellement la greffe de cellules souches allogéniques. Bien que des rémissions à long terme puissent être observées sous ce traitement, il ne faut pas sous-estimer l’augmentation de la mortalité et de la morbidité associées à la transplantation [16,20]. Malgré tous les progrès scientifiques, le traitement du syndrome de Sézary, hormis la greffe de cellules souches, reste à ce jour palliatif.
Malgré le mauvais pronostic actuel de cette maladie, on peut espérer que les découvertes récentes concernant le syndrome de Sézary en tant que pathologie à part entière et les progrès diagnostiques et thérapeutiques qui en découlent permettront d’améliorer les chances de guérison et la qualité de vie des personnes touchées.
Messages Take-Home
- Seuls 25,5% des patients atteints du syndrome de Sézary présentent une érythrodermie classique comme première manifestation.
- L’implication sanguine constitue un élément essentiel du syndrome de Sézary et le distingue des autres lymphomes cutanés primaires.
- Les symptômes non spécifiques de la maladie aux stades initiaux entraînent une latence diagnostique évidente.
- La photophérèse extracorporelle et les nouvelles thérapies ciblées (par ex. au moyen d’anticorps monoclonaux humanisés) améliorent de plus en plus le succès thérapeutique du syndrome de Sézary.
- La transplantation de cellules souches allogéniques est actuellement la seule option de traitement ayant un potentiel curatif.
Littérature :
- Swerdlow SH, Campo E, et al : The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016 ; 127(20) : 2375-2390.
- Scarisbrick JJ, Prince HM, et al : Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome : Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J Clin Oncol 2015 ; 33(32) : 3766-3773.
- Saulite I, Hoetzenecker W, et al : Syndrome de Sézary et dermatite atopique : Comparaison des aspects immunologiques et des cibles. Biomed Res Int 2016 ; article id : 9717530.
- Bradford PT, SS D, et al : Cutaneous lymphoma incidence patterns in the United States : a population-based study of 3884 cas. Blood 2009 ; 113(21) : 5064-5073.
- Wilson LD, Hinds G, Yu JB : Age, race, sexe, stade, et incidence du lymphome cutané. Clin Lymphoma Myeloma Leuk 2012 ; 12(5) : 291-296.
- Vonderheid EC, Bernengo MG, et al : Update on erythrodermic cutaneous T-cell lymphoma : report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 2002 ; 46(1) : 95-106.
- Mangold AR, Thompson AK et al : Manifestations cliniques précoces du syndrome de Sézary : une étude de cohorte multicentrique rétrospective. J Am Acad Dermatol 2017 ; pii : S0190-9622(17) : 31784-X. doi : 10.1016/j.jaad.2017.05.036. [Epub ahead of print].
- Foss FM, Girardi M : Mycosis fongoïde et syndrome de Sézary. Hematol Oncol Clin North Am 2017 ; 31(2) : 297-315.
- Stevens SR, Ke MS, et al : Quantifying skin disease burden in mycosis fungoides-type cutaneous T-cell lymphomas : the severity-weighted assessment tool (SWAT). Arch Dermatol 2002 ; 138(1) : 42-48.
- Trotter MJ, Whittaker SJ, Orchard GE, Smith NP : histopathologie cutanée du syndrome de Sézary : une étude de 41 cas avec un clone de cellules T circulantes prouvé. J Cutan Pathol 1997 ; 24(5) : 286-291.
- Boonk SE, Zoutman WH, et al. : Évaluation des biomarqueurs immunophénotypiques et moléculaires du syndrome de Sézary à l’aide de procédures opérationnelles standard : A Multicenter Study of 59 Patients. J Invest Dermatol 2016 ; 136(7) : 1364-1372.
- Gibson JF, Huang J et al : Lymphome cutané à cellules T (CTCL) : pratiques actuelles en matière d’évaluation sanguine et l’utilité de la restriction de la chaîne Vbeta du récepteur des cellules T (TCR). J Am Acad Dermatol 2016 ; 74(5) : 870-877.
- Lukowsky A, Muche JM et al : Evaluation of T-cell clonality in archive skin biopsy samples of cutaneous T-cell lymphoma using the biomed-2 PCR protocol. Diagn Mol Pathol 2010 ; 19(2) : 70-77.
- Guenova E, Ignatova D et al. : Expression de CD164 sur les cellules T malignes dans le syndrome de Sézary. Acta Derm Venereol 2016 ; 96(4) : 464-467.
- Benoit BM, Jariwala N, et al : CD164 identifie les cellules T CD4+ hautement exprimantes des gènes associés à la malignité dans le syndrome de Sézary : les gènes de signature de Sézary, FCRL3, Tox, et miR-214. Arch Dermatol Res 2017 ; 309(1) : 11-19.
- Jawed SI, Myskowski PL, et al. : Lymphome cutané primaire à cellules T (mycosis fongoïde et syndrome de Sézary) : partie II. Prognosis, management, and future directions. J Am Acad Dermatol 2014 ; 70(2) : 223.e1-17.
- Scarisbrick JJ, Kim YH, et al : Prognostic factors, prognostic indices and staging in mycosis fungoides and Sézary syndrome : where are we now ? Br J Dermatol 2014 ; 170(6) : 1226-1236.
- Olsen E, Vonderheid E, Pimpinelli N, et al : Révisions de la classification du mycosis fongoïde et du syndrome de Sézary : une proposition de l’International Society for Cutaneous Lymphomas (ISCL) et du groupe de travail sur les lymphomes cutanés de l’Organisation européenne de recherche et de traitement du cancer (EORTC). Blood 2007 ; 110(6) : 1713-1722.
- Dippel E, Assaf C, Becker J, Bergwelt M, Beyer M : S2k – Kurzleitlinie – Kutane Lymphome (ICD10 C82 – C86) Update 2016. JDDG 2017 ; [in Vorbereitung].
- Trautinger F, Eder J, et al : European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome – Update 2017. Eur J Cancer 2017 ; 77 : 57-74.
- DeSimone JA, Sodha P, Ignatova D, et al : Recent advances in primary cutaneous T-cell lymphoma. Curr Opin Oncol 2015 ; 27(2) : 128-133.
- Izykowska K, Przybylski GK, et al : Les réarrangements génétiques entraînent une altération de l’expression génétique et de nouveaux transcrits de fusion dans le syndrome de Sézary. Oncotarget 2017 ; 8(24) : 39627-39639.
- Guenova E, Watanabe R et al : TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res 2013 ; 19(14) : 3755-3763.
- Campbell JJ, Clark RA, et al : Le syndrome de Sézary et le mycosis fongoïde proviennent de sous-ensembles distincts de cellules T : une raison biologique pour leurs comportements cliniques distincts. Blood 2010 ; 116(5) : 767-771.
- Willemze et al : La mise à jour 2018 de la classification WHO-EORTC pour les lymphomes cutanés primaires, Blood (2019) 133 (16) : 1703-1714.
- Dippel E, et al. : S2k-Leitlinie – Kutane Lymphome Update 2016 – Teil 2 : Therapie und Nachsorge (ICD10 C82 – C86), J Dtsch Dermatol Ges. 2018 Jan;16(1) : 112-123.
- Lundin J, Hagberg H, Repp R, et al. Étude de phase 2 de l’alemtuzumab (anticorps monoclonal anti-CD52) chez des patients atteints de mycosis fongoïde/ syndrome de Sézary en phase avancée. Blood. 2003;101 : 4267-4272.
- Bernengo MG, Quaglino P, Comessatti A, et al. : Low-dose intermittent alemtuzumab in the treatment of Sezary syndrome : clinical and immunologic findings in 14 patients. Haematologica 2007 ; 92 : 784-794.
- Kim YH, et al : Mogamulizumab vs vorinostat dans le lymphome cutané à cellules T précédemment traité (MAVORIC) : un essai international, en ouvert, randomisé, contrôlé de phase 3. Lancet Oncol. 2018;19 : 1192-1204).
- Phillips T, Devata S, Wilcox RA : Challenges and opportunities for checkpoint blockade in T-cell lymphoproliferative disorders. J Immunother Cancer 2016 ; 4 : 95.
DERMATOLOGIE PRATIQUE 2019 ; 29(6) : 14-18









