Les pneumopathies interstitielles sont relativement rares dans la pratique de la médecine générale. Il faut néanmoins y penser en cas de dyspnée chronique, de toux sèche et de râles crépitants à fines bulles. Un diagnostic précoce et correct est essentiel.
Les pneumopathies interstitielles (PID) regroupent un grand nombre de maladies rares et différentes du parenchyme pulmonaire qui ont une présentation clinique, radiologique et pathologique similaire [1]. Les processus sous-jacents à la DLI, le pronostic, mais aussi les traitements diffèrent considérablement d’une entité à l’autre. Nous trouvons d’une part des maladies avec un pronostic favorable, mais aussi des maladies avec une évolution aiguë et une mortalité élevée. La fibrose pulmonaire idiopathique (FPI), la plus fréquente des DLI parmi les pneumopathies interstitielles idiopathiques (PII), présente une évolution comparable à celle des tumeurs malignes agressives, avec une survie moyenne d’environ trois ans après le diagnostic [2].
Le traitement de la DLI chez un patient donné dépend essentiellement de l’entité de la maladie. Pour certaines pathologies, l’éviction de l’exposition à des toxiques (inhalés) est centrale. Selon l’entité, des traitements immunosuppresseurs ou, dans le cas de la FPI, un traitement antifibrotique sont utilisés. Un diagnostic précoce et correct, avec notamment l’identification des facteurs réversibles, est essentiel pour conseiller et traiter au mieux les patients.
Classification des pneumopathies interstitielles
Les ILD peuvent être classées en maladies dont la cause est connue et en maladies dont la cause est inconnue (pneumopathies interstitielles idiopathiques, PII) (Fig. 1). La liste des causes possibles des ILD est longue. Parmi les causes connues, nous trouvons des expositions à des poussières inorganiques ou organiques, à des médicaments et à des rayons X. Les causes les plus fréquentes sont les suivantes De nombreuses maladies rhumatologiques peuvent se compliquer d’une DLI. L’ILD peut être la première manifestation d’une maladie rhumatologique sous-jacente avant que celle-ci ne puisse être détectée à l’aide des critères diagnostiques habituels. Pour tenir compte de cette situation, le terme “interstitial pneumonitis with autoimmune features” (IPAF) a été introduit en 2015 [3].
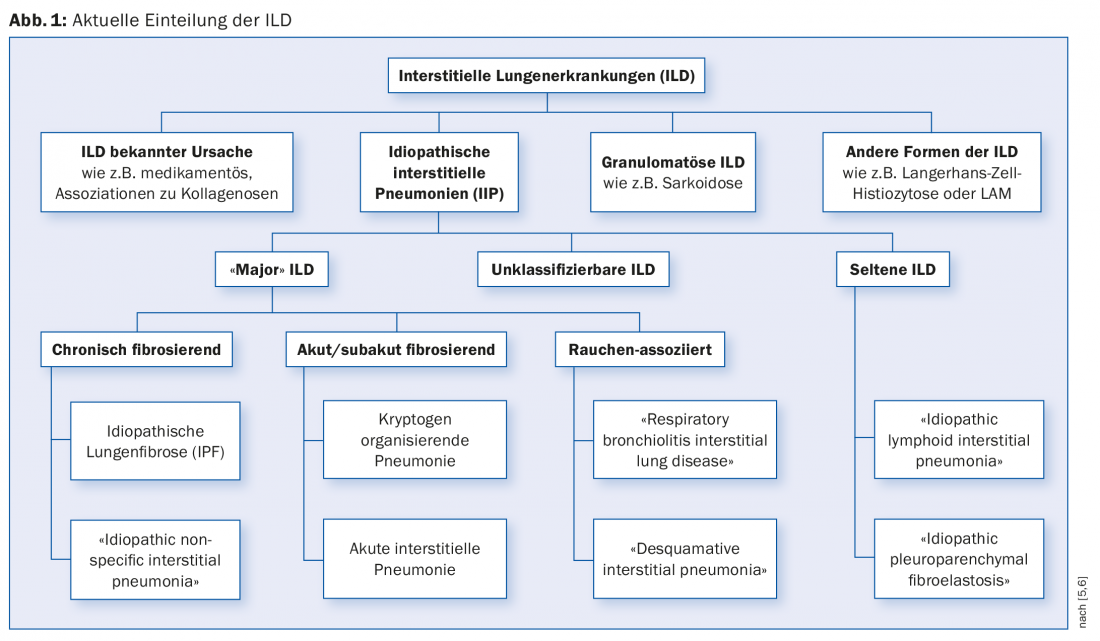
Comme son nom l’indique, la cause de l’IIP n’est pas claire. Les IIP “majeures” sont classées en fonction de leur évolution clinique : fibrosantes chroniques, fibrosantes aiguës/subaiguës et associées au tabagisme.
Pour le groupe des maladies associées au tabagisme, le terme “idiopathique” est parfois en contradiction avec la physiopathologie, une catégorisation en dehors de l’IIP est donc discutée. Le tabagisme provoque des modifications histologiquement détectables (“bronchiolite respiratoire”). Les individus sensibles peuvent développer une ILD (“respiratory bronchiolitis interstitial lung disease” [RB-ILD] et “desquamative interstitial pneumonia”) [4].
Épidémiologie de l’ILD
Les ILD sont des maladies rares, par conséquent, il existe peu de données épidémiologiques. Une grande étude épidémiologique réalisée en 1994 a révélé une incidence de 31,5 cas pour 100 000/an chez les hommes et de 26,1 cas pour 100 000/an chez les femmes [7]. La FPI, qui touche plus fréquemment les hommes âgés, est l’entité la plus fréquente parmi les IILD. Il n’existe pas de données épidémiologiques sur la FPI en Suisse. On estime que 100 à 5000 patients souffrent de FPI en Suisse (prévalence de 1,25 à 63 cas/100 000) [8]. L’âge au moment du premier diagnostic de la DLI varie entre les différentes entités de la maladie. Alors que l’incidence de la FPI augmente nettement avec l’âge, les sarcoïdoses, les histiocytoses à cellules de Langerhans ou, chez les femmes, les lymphangioléiomyomatoses (LAM) sont plus fréquentes chez les jeunes adultes.
Diagnostic de l’ILD
La DLI est diagnostiquée en tenant compte des résultats cliniques, radiologiques, fonctionnels pulmonaires, chimiques de laboratoire et, selon la situation, cytologiques/histologiques. La recherche du diagnostic et la définition de la stratégie thérapeutique pour chaque patient se font de manière optimale dans un cadre interdisciplinaire composé de cliniciens (pneumologues), de radiologues et de pathologistes (conseil d’administration de l’ILD). Il a été démontré que cela permettait d’améliorer considérablement la qualité du diagnostic [9].
Clinique : La clinique de l’ILD est souvent non spécifique, avec une dyspnée qui augmente lentement et une toux généralement sèche. L’examen clinique révèle souvent des crépitants à fines bulles accentués à la base, ainsi qu’une diminution de la saturation en oxygène (surtout à l’effort) et des signes d’hypoxémie chronique tels que des ongles en verre de montre et des doigts en baguettes de tambour, surtout dans les cas avancés. Il est important de penser rapidement à la présence d’une DLI dans le diagnostic différentiel des patients présentant ces symptômes.
Souvent, une anamnèse minutieuse nous fournit des indices décisifs sur l’entité de l’ILD. La durée des symptômes, mais aussi les éventuelles influences environnementales, y compris une anamnèse professionnelle détaillée, sont importantes. L’identification des influences environnementales potentiellement réversibles est très importante. L’identification d’éventuels aéroallergènes susceptibles de déclencher une pneumonie d’hypersensibilité (alvéolite allergique exogène, AAE) est fondamentale, tant pour l’établissement du diagnostic que pour l’éviction décisive de l’exposition. Nous trouvons le plus souvent un élevage d’oiseaux, un contact avec l’agriculture ou une exposition aux moisissures. Le tabagisme est un facteur déclenchant potentiellement réversible dans les DLI associées au tabagisme. Outre la pleurésie liée à l’amiante, l’exposition à l’amiante peut également entraîner une fibrose pulmonaire liée à l’amiante. Les symptômes d’une maladie rhumatologique sous-jacente doivent être recherchés de manière ciblée. De nombreux médicaments peuvent avoir pour effet secondaire de déclencher une ILD. Nous souhaitons attirer l’attention sur le site www.pneumotox.com, très utile au quotidien, qui fournit des informations sur les effets secondaires connus de la toxicité pulmonaire.
L’anamnèse familiale doit être recherchée, jusqu’à 20% des cas d’ILD présentent une forme familiale avec des mutations génétiques correspondantes [10].
Imagerie : la radiographie conventionnelle du thorax permet souvent de deviner une pneumopathie interstitielle en cas d’augmentation du dessin réticulaire et/ou nodulaire des poumons.
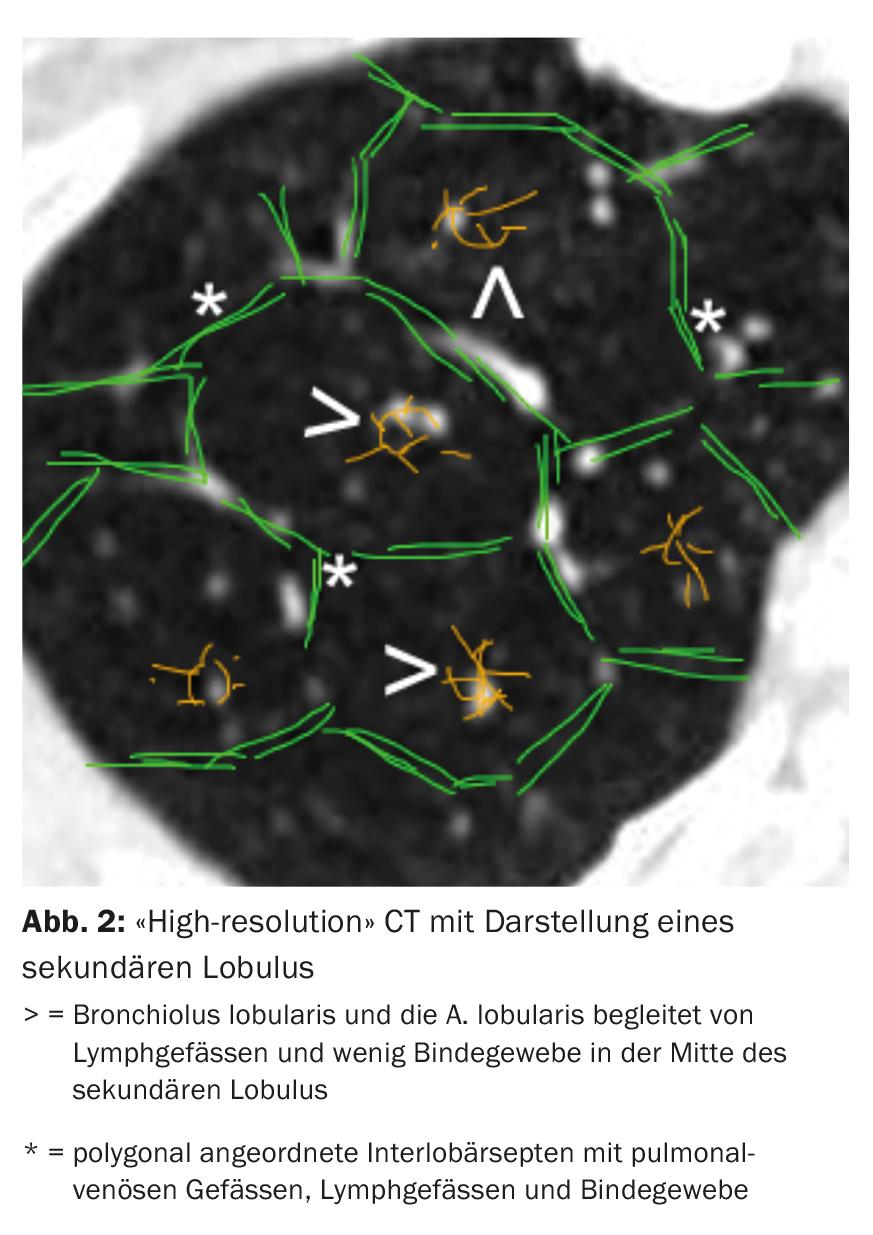
La tomodensitométrie (TDM) du thorax avec des coupes “haute résolution” occupe une place centrale dans le diagnostic de la DLI. Différents schémas radiologiques peuvent être distingués en fonction du type et de la disposition des modifications par rapport à la localisation du lobule secondaire (Fig. 2) et à la topographie des poumons.
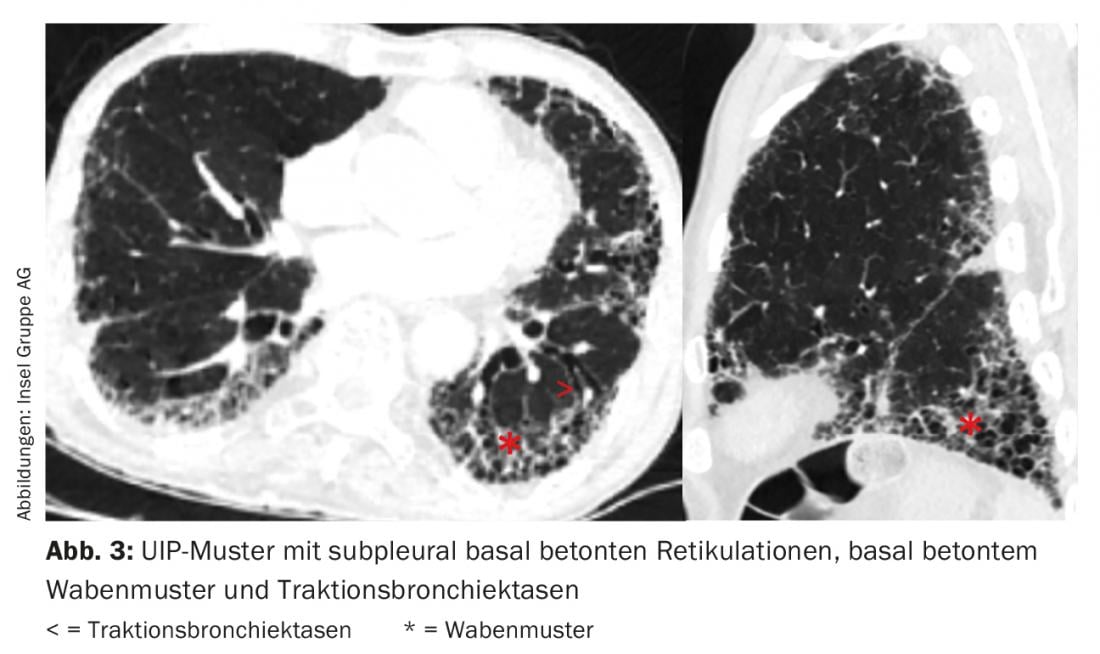
Le modèle radiologique unique n’est pas spécifique à une entité de la DLI. Par exemple, dans la FPI, nous trouvons un modèle de “pneumonie interstitielle habituelle” (UIP) avec des réticulations à accentuation basale sous-pleurale, un modèle en nid d’abeille à accentuation basale et des bronchectasies par traction (Fig. 3) [11]. Cependant, un modèle UIP peut également être trouvé dans d’autres entités telles qu’une vascularite des petits vaisseaux, une EAA chronique, une ILD dans le cadre d’une polyarthrite rhumatoïde ou une asbestose.
Fonction pulmonaire : dans les DLI, nous trouvons généralement une restriction et une altération des échanges gazeux (capacité de diffusion). Au fur et à mesure de la progression de la maladie, une hypoxémie d’effort apparaît, suivie d’une hypoxémie de repos.
La capacité vitale forcée (CVF) est utilisée comme un paramètre évolutif important. Une chute de la CVF de ≥10% en 24 semaines multiplie par 4,8 le risque de mortalité pour les douze mois suivants [12]. Outre la FVC, facile à mesurer, la capacité de diffusion, le test de marche de 6 minutes et les indices multivariables sont également en corrélation avec le pronostic [13].
Laboratoire : Le laboratoire peut être utile pour la classification de l’ILD. En plus de l’hémogramme (éosinophilie), la fonction rénale, les tests hépatiques et inflammatoires ainsi que la recherche sérologique d’une maladie rhumatologique sous-jacente peuvent nous donner des indications.
Examens invasifs : En règle générale, une bronchoscopie avec au moins un lavage broncho-alvéolaire (LBA) est réalisée chez les patients chez qui une DLI vient d’être diagnostiquée. Un LBA de plus en plus hémorragique d’une portion à l’autre est typique d’une hémorragie alvéolaire (par exemple, dans les vascularites des petits vaisseaux). La distribution des cellules dans le LBA présente des changements caractéristiques dans certaines entités de la DLI (par exemple, l’éosinophilie dans la pneumonie à éosinophiles, la lymphocytose dans la sarcoïdose et l’EAA). En fonction du diagnostic de suspicion basé sur la présentation clinique et radiologique, des biopsies ganglionnaires médiastinales/hilaires et/ou des biospies pulmonaires transbronchiques sont réalisées. Les fragments de tissu des biopsies pulmonaires transbronchiques sont souvent trop petits pour permettre un diagnostic fiable. Des échantillons de tissus plus importants peuvent être obtenus par thoracoscopie ou, depuis quelques années, par bronchoscopie avec cryobiopsies.
Thérapie
Le traitement de la DLI diffère fondamentalement d’une entité à l’autre (Fig. 4). Ainsi, pour un petit nombre d’entités, il est possible d’influer de manière décisive sur la maladie par une éviction de l’exposition (arrêt du tabac, éviction des déclencheurs d’une alvéolite allergique exogène, changement de médicaments). De nombreuses DLI sont traitées par immunosuppression, comme les maladies associées aux collagénoses/vascularites, les pneumonies organisatrices (cryptogéniques), les pneumonies à éosinophiles ou les sarcoïdoses. L’immunosuppression est généralement effectuée à l’aide de corticostéroïdes systémiques, souvent en combinaison avec un médicament épargnant les stéroïdes (par exemple l’azathioprine ou le mycophénolate).
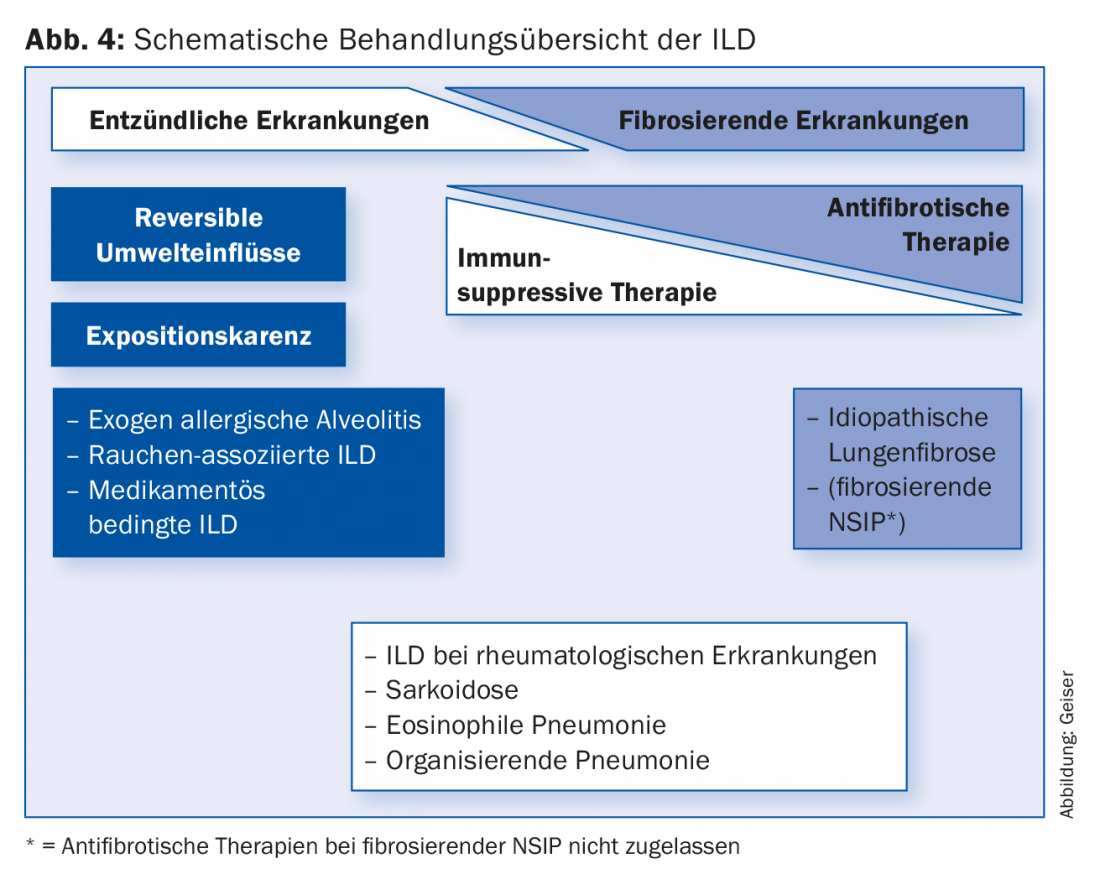
Les traitements immunosuppresseurs utilisés dans la FPI jusqu’à il y a quelques années ne sont plus recommandés [14,15]. Après de nombreuses études négatives avec diverses substances, les deux médicaments actuellement autorisés pour le traitement de la FPI, la pirfénidone (Esbriet®) et le nintedanib (Ofev®), ont permis de montrer un ralentissement de la progression de la maladie [16–18]. De nouvelles données indiquent que la survie pourrait être favorablement influencée.
Chez les jeunes patients atteints d’entités de la DLI à progression rapide, comme la FPI, une évaluation de la transplantation pulmonaire devrait être effectuée tôt dans l’évolution de la maladie. Au fur et à mesure de la progression de la DLI, une oxygénothérapie de longue durée peut être nécessaire chez les patients hypoxémiques et d’autres traitements palliatifs, y compris la réadaptation pulmonaire.
Messages Take-Home
- Bien que les pneumopathies interstitielles (PID) soient relativement rares dans la pratique de la médecine générale, il convient de les traiter en cas de dyspnée chronique, de sécheresse de l’haleine et d’autres symptômes.
- Il faut y penser en cas de toux et de crépitants à fines bulles.
- Parmi les ILD, nous trouvons des entités pathologiques très différentes avec des évolutions différentes.
- Un diagnostic précoce et correct est essentiel pour conseiller et traiter au mieux les patients.
- Le traitement de la DLI varie considérablement en fonction de l’entité pathologique et va de l’éviction de l’exposition aux agents déclencheurs à l’utilisation de traitements antifibrotiques dans la fibrose pulmonaire idiopathique (FPI), en passant par l’immunosuppression.
- Les patients plus jeunes présentant une entité DLI à progression rapide doivent être évalués précocement en vue d’une transplantation pulmonaire.
- Au cours de l’évolution, une oxygénothérapie de longue durée peut être nécessaire chez les patients hypoxémiques et d’autres traitements palliatifs peuvent être administrés.
Littérature :
- Cosgrove G, Schwarz M : Approche de l’évaluation et du diagnostic de la maladie pulmonaire interstitielle. In : Schwarz M, King T (éd.) : Maladie pulmonaire interstitielle. 5e éd. Shelton : People’s Medical Publishing House-USA 2011 : 3-34.
- Latsi PI, et al : Fibrotic idiopathic interstitial pneumonia : the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003 ; 168(5) : 531-537.
- Fischer A, et al : An official European Respiratory Society/American Thoracic Society research statement : interstitial pneumonia with autoimmune features. Eur Respir J 2015 ; 46(4) : 976-987.
- Fraig M, et al : Respiratory bronchiolitis : a clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am J Surg Pathol 2002 ; 26(5) : 647-653.
- American Thoracic Society/European Respiratory Society : International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002 ; 165(2) : 277-304.
- Travis WD, et al : An official American Thoracic Society/European Respiratory Society statement : Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013 ; 188(6) : 733-748.
- Coultas DB, et al : The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 1994 ; 150(4) : 967-972.
- Funke M, Geiser T : Fibrose pulmonaire idiopathique : le point de basculement est maintenant ! Swiss Med Wkly 2015 ; 145 : w14139.
- Flaherty KR, et al : Pneumonie interstitielle idiopathique : quel est l’effet d’une approche multidisciplinaire du diagnostic ? Am J Respir Crit Care Med 2004 ; 170(8) : 904-910.
- Kropski JA, Blackwell TS, Loyd JE : The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J 2015 ; 45(6) : 1717-1727.
- Raghu G, et al : An official ATS/ERS/JRS/ALAT statement : idiopathic pulmonary fibrosis : evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011 ; 183(6) : 788-824.
- du Bois RM, et al : Forced vital capacity in patients with idiopathic pulmonary fibrosis : test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011 ; 184(12) : 1382-1389.
- Sharp C, Adamali HI, Millar AB : A comparison of published multidimensional indices to predict outcome in idiopathic pulmonary fibrosis. ERJ Open Res 2017 ; 3(1). DOI: 10.1183/23120541.00096-2016.
- Funke M, et al. : Fibrose pulmonaire idiopathique en Suisse : diagnostic et traitement. Respiration 2017 ; 93(5) : 363-378.
- Raghu G, et al : An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline : Treatment of Idiopathic Pulmonary Fibrosis (Traitement de la fibrose pulmonaire idiopathique). Une mise à jour du guide de pratique clinique 2011. Am J Respir Crit Care Med 2015 ; 192(2) : e3-19.
- Noble PW, et al : Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY) : two randomised trials. Lancet 2011 ; 377(9779) : 1760-1769.
- Richeldi L, et al : Efficacité et sécurité du nintedanib dans la fibrose pulmonaire idiopathique. N Engl J Med 2014 ; 370(22) : 2071-2082.
- King TE Jr, et al : A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014 ; 370(22) : 2083-2092.
PRATIQUE DU MÉDECIN DE FAMILLE 2017 ; 12(12) : 13-18