Les paranodopathies sont une nouvelle forme particulière de polyneuropathies inflammatoires, médiées par des lésions de l’anneau de Ranvier induites par des auto-anticorps. Quelles sont les caractéristiques de cette maladie et comment peut-on la traiter ?
Les polyneuropathies sont un groupe hétérogène de maladies qui résultent de l’endommagement de l’axone ou de la gaine de myéline des nerfs périphériques. Même si la cause de 25% des polyneuropathies n’est pas claire, la différenciation étiologique est essentielle dans la pratique clinique quotidienne [1,2]. Cela permet d’identifier les causes traitables et de proposer des thérapies spécifiques, notamment pour les neuropathies immunitaires. Le diagnostic de base, outre l’anamnèse, l’examen et le diagnostic de laboratoire, comprend l’électroneurographie, qui permet de distinguer un schéma axonal d’un schéma démyélinisant, et l’échographie nerveuse. Ces dernières années, une nouvelle entité a été décrite, les paranodopathies, qui ne peuvent être classées selon les concepts classiques d'”axonal” et de “démyélinisant” [3–5]: ici, l’anneau de laçage de Ranvier représente le point de départ de la maladie. Les auto-anticorps paranodaux sont des biomarqueurs précieux dans les neuropathies immunitaires, car les paranodopathies diffèrent des autres neuropathies immunitaires en termes de réponse au traitement.
Neuropathies inflammatoires
Les neuropathies inflammatoires comprennent, outre les neuropathies infectieuses rares en Europe centrale, les neuropathies à médiation auto-immune qui, avec une prévalence d’environ 10%, représentent la troisième cause la plus fréquente de toutes les polyneuropathies, après les polyneuropathies diabétiques et éthyliques [2,6]. Les représentants éminents des formes démyélinisantes sont le syndrome de Guillain-Barré (SGB) à évolution aiguë, la polyneuropathie inflammatoire démyélinisante chronique (PIDC) et la neuropathie motrice multifocale (NMM). Dans ces maladies, la gaine de myéline est endommagée par des processus immunitaires humoraux et cellulaires, et une dégénérescence axonale peut se produire secondairement. En revanche, la neuropathie vasculaire se caractérise par une lésion axonale ischémique due à une atteinte généralement isolée des vasa nervorum. L’évolution, le schéma de distribution, le diagnostic de laboratoire et surtout l’électrophysiologie et la biopsie nerveuse sont déterminants pour le diagnostic différentiel des neuropathies immunitaires. La différenciation précise est particulièrement importante en ce qui concerne les différentes options thérapeutiques (tableau 1).
Le paranoïde, point de départ de neuropathies immunitaires rares
L’anneau de Ranvier des nerfs périphériques myélinisés est divisé en trois sections (Fig. 1). Au niveau du nodium, les canaux sodiques voltage-dépendants, responsables de la transmission saltatoire de l’excitation, sont regroupés par la neurofascine-186 [7]. Cette région est flanquée du paranoïde, où les boucles de myéline terminales se trouvent de manière lâche autour de l’axone. Le complexe protéique de la neurofascine-155 gliale et de la contactine-1 et caspr-1 axonales relie ici la myéline et l’axone et joue un rôle essentiel dans le maintien de l’architecture nodale et des vitesses normales de conduction nerveuse [8,9]. Latéralement, sur le juxtaparanodium, des canaux potassiques voltage-dépendants sont concentrés par un complexe protéique de contactine-2 et de caspr-2 [10].
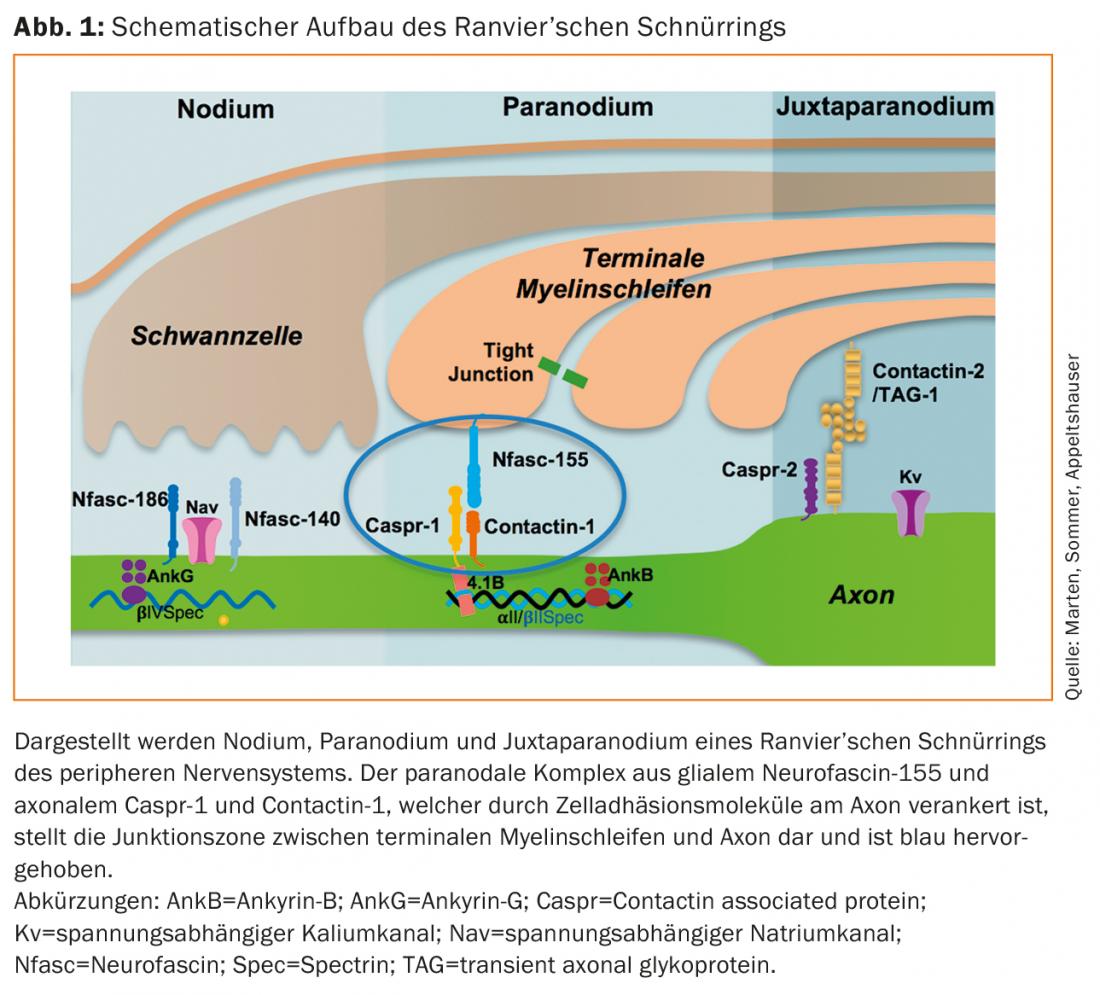
Plusieurs études histopathologiques décrivent une altération de l’architecture des anneaux de laçage dans différentes polyneuropathies [11–13]. Ces dernières années, des autoanticorps IgG et plus récemment des autoanticorps IgM contre les protéines paranodales neurofascine-155, contactine-1 et caspr-1 ont été décrits chez des patients atteints de neuropathies immunitaires [14–20]. La plupart du temps, il s’agit d’autoanticorps de la sous-classe 4 des IgG, indépendants du complément, mais des autoanticorps IgG3 ont été décrits chez des patients au stade aigu de la maladie, capables de lier et d’activer le complément in vitro [16,21]. Sur le plan électrophysiologique, ces neuropathies immunitaires médiées par des auto-anticorps peuvent répondre aux critères d’une PIDC ou d’un SGB, mais pas sur le plan histopathologique. Dans ce cas, il n’y a pas de dé- et de remyélinisation dépendante des macrophages, mais une lésion axonale (secondaire) et un détachement des boucles de myéline terminales de l’axone paranodal, ainsi qu’un allongement des nodosités et une dispersion des protéines et des canaux ioniques paranodaux [14,16,22,23]. On pense que cette anomalie architecturale de l’anneau de Ranvier est responsable de blocs de conduction et d’un allongement de la vitesse de conduction nerveuse dans les paranodopathies [5,24]. Des modèles de transfert passif confirment une destruction paranodale et des blocs de conduction in vivo après l’injection d’autoanticorps IgG4 anti-contactine-1 et IgG anti-neurofascine-155 [25,26]. Le point de départ de la maladie n’est donc pas la gaine de myéline, mais l’anneau de laçage, ce qui explique pourquoi il n’est pas possible de classer la maladie dans les catégories classiques “axonale” ou “démyélinisante”. C’est pourquoi le terme “paranodopathie”, qui se référait à l’origine aux neuropathies immunitaires associées aux auto-anticorps anti-gangliosides, a été établi pour désigner plus particulièrement les neuropathies immunitaires associées aux auto-anticorps paranodaux [3,4,22,27].
Caractéristiques cliniques, diagnostic et traitement
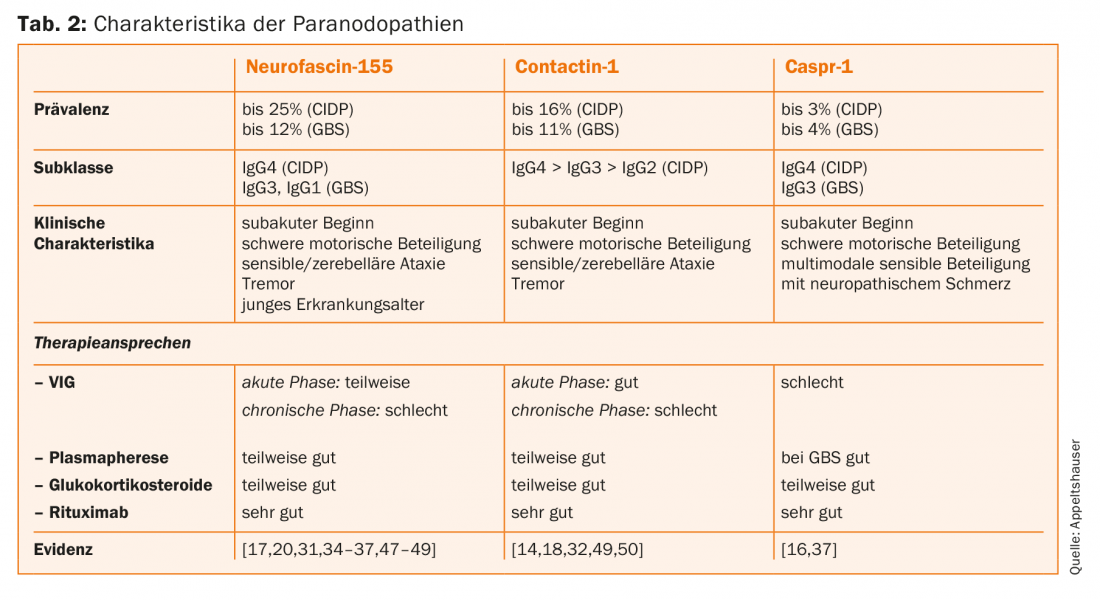
Les auto-anticorps paranodaux ont jusqu’à présent été identifiés presque exclusivement chez des patients présentant un tableau clinique de PIDC et de SGB. Les prévalences sont généralement de <10% [28–30]. Les patients atteints présentent un début de maladie aigu ou subaigu et développent une participation motrice sévère. En outre, un tremblement prononcé et une ataxie sensitive peuvent aggraver l’évolution clinique [31,32]. La prévalence la plus élevée est celle des anticorps IgG4 dirigés contre la neurofascine-155 [33]. Chez ces patients séropositifs, des foyers de démyélinisation centrale, des tremblements et une ataxie cérébelleuse ont également été décrits comme des signes possibles de lésions médiées par des auto-anticorps centraux. Le pathomécanisme et la prévalence des auto-anticorps IgG anti-neurofascine-155 chez les patients présentant une atteinte centrale et périphérique cliniquement combinée n’ont pas encore été suffisamment étudiés et font l’objet de controverses dans la littérature [30,33–36]. Des auto-anticorps anti-Caspr-1 ont rarement été détectés, mais des auto-anticorps IgG4 ont été décrits chez des patients atteints d’une maladie chronique et des auto-anticorps IgG3 chez une patiente atteinte du SGB [16,37]. La douleur neuropathique a été proposée comme caractéristique commune avec un corrélat histopathologique, à savoir une liaison des auto-anticorps aux neurones nociceptifs des ganglions rachidiens. Cependant, il n’existe pas d’études cliniques et expérimentales de grande envergure pour le confirmer [16]. Le tableau 2 donne un aperçu des caractéristiques spécifiques aux autoanticorps des paranodopathies.
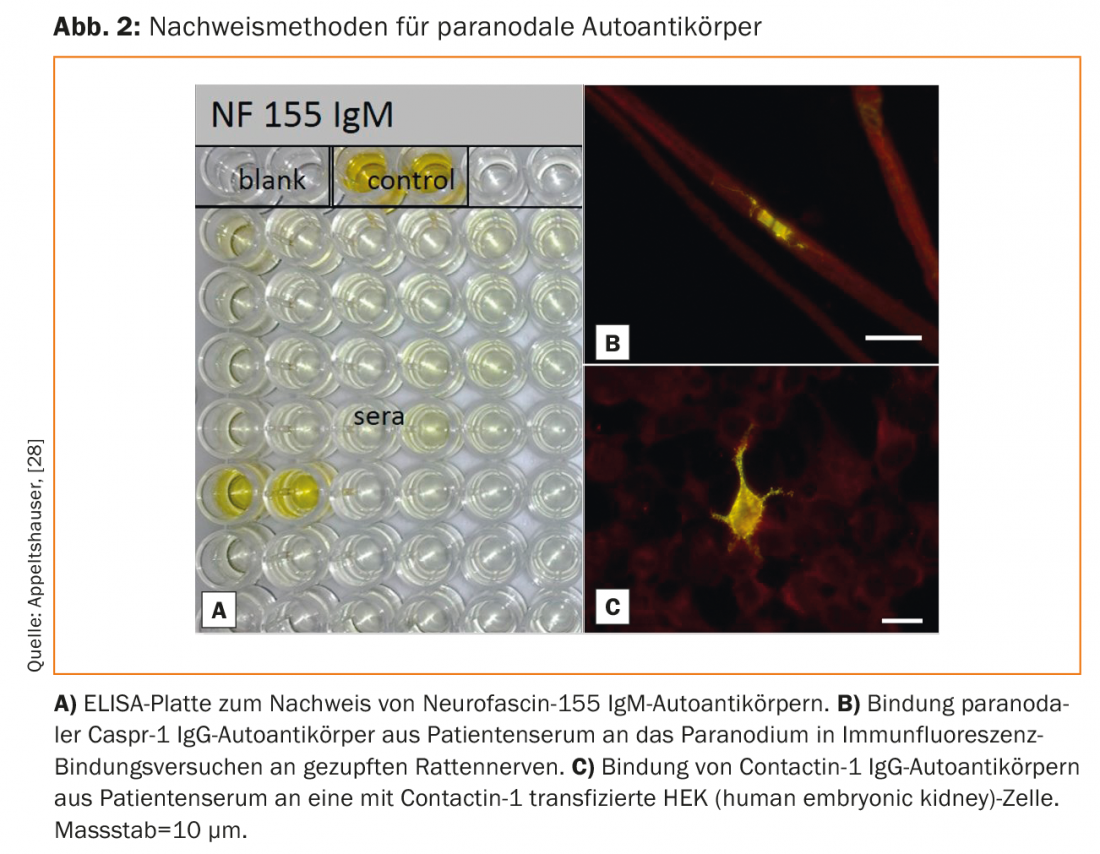
Les autoanticorps peuvent être détectés dans le sérum par ELISA et par des tests de liaison sur des nerfs pincés de souris ou de rats et sur des cellules transfectées (Fig. 2) [33]. Il n’existe pas encore de test commercial validé, mais il est possible de le tester dans des laboratoires de recherche spécialisés. Le dépistage n’est utile que pour les patients présentant un phénotype clinique typique. En revanche, dans le cas de neuropathies sensitivomotrices légères, chroniques et à prédominance distale, il est préférable de procéder à une évaluation des causes fréquentes.
En raison du faible nombre de cas de patients atteints de paranodopathies, il n’existe pas encore d’études thérapeutiques contrôlées. Des séries de cas indiquent que, contrairement au CIDP classique, les patients atteints de paranodopathie à médiation IgG4 répondent mal aux IgIV, alors que les patients atteints de paranodopathie à médiation IgG3 y répondent bien [14,21,30].
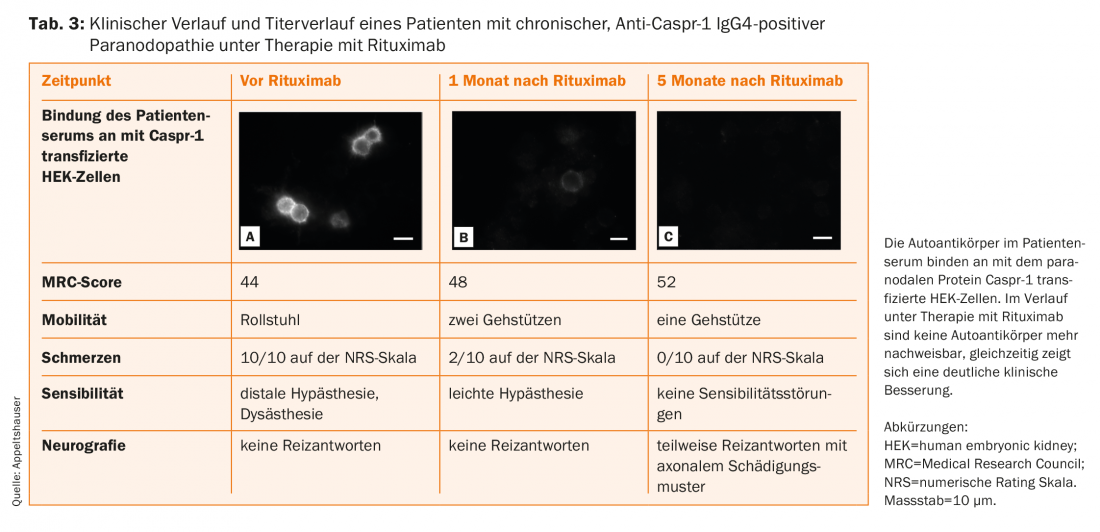
Dans les séries de cas, les patients présentant des autoanticorps IgG4 répondent très bien au rituximab, avec une réduction significative des titres d’anticorps comme paramètre de substitution ( tableau 3) [14,16,20,38]. Il est possible qu’un début de traitement précoce permette d’éviter une lésion axonale chronique. C’est pourquoi, en cas de détection d’auto-anticorps IgG4 paranodaux, la ligne directrice actuelle recommande également un essai thérapeutique individuel avec le rituximab, qui doit être effectué dans des centres spécialisés avec un suivi étroit [39]. En outre, le suivi des patients atteints de paranodopathies est essentiel pour mieux comprendre la maladie de cette nouvelle entité.
Messages Take-Home
- L’étiologie des polyneuropathies inflammatoires est hétérogène et diffère selon qu’il s’agit de polyneuropathies axonales ou démyélinisantes.
- Les paranodopathies sont une nouvelle forme particulière de polyneuropathies inflammatoires, médiées par des lésions de l’anneau de Ranvier induites par des auto-anticorps.
- Les caractéristiques cliniques des paranodopathies sont : un début subaigu, une atteinte motrice sévère, une mauvaise réponse aux traitements standards, des tremblements et un jeune âge de survenue de la maladie pour les autoanticorps anti-Neurofascine-155, avec en plus une ataxie pour les autoanticorps anti-Contactine-1 et des douleurs pour les anticorps anti-Caspr-1.
- L’identification des patients présentant des auto-anticorps paranodaux est importante pour une meilleure compréhension de la maladie et pour la prise de décisions thérapeutiques.
Littérature :
- Pasnoor M, et al : Polyneuropathie sensorielle cryptogénique. Neurol Clin 2013 ; 31(2) : 463-476.
- Visser NA, et al. : Incidence de la polyneuropathie à Utrecht, aux Pays-Bas. Neurology 2015 ; 84(3) : 259-264.
- Uncini A : Un mécanisme commun et une nouvelle catégorisation pour les neuropathies médiées par les anticorps anti-gangliosides. Exp Neurol 2012 ; 235(2) : 513-516.
- Uncini A, Vallat JM : Nodo-paranodopathies auto-immunes du nerf périphérique : le concept gagne du terrain. J Neurol Neurosurg Psychiatry 2017 ; 89(6) : 627-635.
- Uncini A, Susuki K, Yuki N : Nodo-paranodopathy : au-delà de la classification démyélinisante et axonale dans les neuropathies médiées par les anticorps anti-gangliosides. Clin Neurophysiol 2013 ; 124(10) : 1928-1934.
- Hanewinckel R, et al : Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology 2016 ; 87(18) : 1892-1898.
- Stathopoulos P, Alexopoulos H, Dalakas MC : Cibles antigéniques auto-immunes au niveau du nœud de Ranvier dans les troubles démyélinisants. Nat Rev Neurol 2015 ; 11(3) : 143-156.
- Vallat JM, et al : Contactin-Associated Protein 1 (CNTNAP1) Mutations Induce Characteristic Lesions of the Paranodal Region. J Neuropathol Exp Neurol 2016 ; 75(12) : 1155-1159.
- Sherman DL, et al : Les neurofascins sont nécessaires pour établir des domaines axonaux pour la conduction saltatoire. Neuron 2005 ; 48(5) : 737-742.
- Poliak S, et al : Caspr2, un nouveau membre de la superfamille des neurexines, est localisé aux juxtaparanodes des axones myélinisés et s’associe aux canaux K+. Neuron 1999 ; 24(4) : 1037-1047.
- Doppler K, et al : Disruption de l’architecture nodale dans les biopsies de peau de patients atteints de neuropathies démyélinisantes. J Peripher Nerv Syst 2013 ; 18(2) : 168-176.
- Li J, et al : Biopsies de la peau dans les neuropathies liées à la myéline : apporter la pathologie moléculaire au chevet du patient. Brain 2005 ; 128(5) : 1168-1177.
- Cifuentes-Diaz C, et al : Nodes of ranvier and paranodes in chronic acquired neuropathies. PloS one 2011 ; 6(1) : e14533.
- Doppler K, et al : Destruction de l’architecture paranodale dans les neuropathies inflammatoires avec auto-anticorps anti-contactine-1. J Neurol Neurosurg Psychiatry 2015 ; 86(7) : 720-728.
- Devaux JJ, et al : Les antibodies à la gliomédine provoquent une neuropathie démyélinisante périphérique et le démantèlement des ganglions de Ranvier. Am J Pathol 2012 ; 181(4) : 1402-1413.
- Doppler K, et al : Auto-antibodies to contactin-associated protein 1 (Caspr) chez deux patients atteints de neuropathie inflammatoire douloureuse. Brain 2016 ; 139(10) : 2617-2630.
- Ng JK, et al : La neurofascine en tant que cible pour les auto-anticorps dans les neuropathies périphériques. Neurology 2012 ; 79(23) : 2241-2248.
- Devaux JJ, Odaka M, Yuki N : Les protéines nodales sont des antigènes cibles dans le syndrome de Guillain-Barre. J Peripher Nerv Syst 2012 ; 17(1) : 62-71.
- Doppler K, et al : Autoantibodies IgM de la neurofascine-155 chez les patients atteints de neuropathies inflammatoires. J Neurol Neurosurg Psychiatry 2018 ; 89(11) : 1145-1151.
- Burnor E, et al. : Anticorps à la neurofascine dans les neuropathies auto-immunes, génétiques et idiopathiques. Neurology 2018 ; 90(1) : e31-e38.
- Appeltshauser L, et al : Le dépôt de complément induit par la liaison des auto-anticorps anti-contactine-1 est modifié par les immunoglobulines. Exp Neurol 2017 ; 287(1) : 84-90.
- Koike H, et al : Dissection paranodale dans la polyneuropathie inflammatoire démyélinisante chronique avec des anticorps anti-neurofascine-155 et anti-contactine-1. J Neurol Neurosurg Psychiatry 2017 ; 88(6) : 465-473.
- Vallat JM, et al. : Subacute nodopathy with conduction blocks and anti-neurofascin 140/186 antibodies : an ultrastructural study. Brain 2018 ; 141(7) : e56.
- Vallat JM, et al : Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with anti-Neurofascin 155 antibodies. Neuromuscul Disord 2017 ; 27(3) : 290-293.
- Manso C, et al : Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain 2016 ; 139(6) : 1700-1712.
- Yan W, et al : Antibodies to neurofascin exacerbate adoptive transfer experimental autoimmune neuritis. J Neuroimmunol 2014 ; 277(1-2) : 13-17.
- Kuwabara S, Misawa S, Mori M : Nodopathy : polyneuropathie inflammatoire démyélinisante chronique avec anticorps anti-neurofascine 155. J Neurol Neurosurg Psychiatry 2017 ; 88(6) : 459.
- Doppler K, et al : Contactin-1 and Neurofascin-155/-186 Are Not Targets of Auto-Antibodies in Multifocal Motor Neuropathy. PloS one 2015 ; 10(7) : e0134274.
- Doppler K, Sommer C : Nouvelle entité des paranodopathies : une structure cible avec des conséquences thérapeutiques. Akt Neurol 2017 ; 44(3) : 194-199.
- Hu W, et al : Association of neurofascin IgG4 and atypical chronic inflammatory demyelinating polyneuropathy : A systematic review and meta-analysis. Brain Behav 2018 : 8(10) : e1115.
- Querol L, et al : Les anticorps IgG4 contre la neurofascine dans le CIDP sont associés à des tremblements invalidants et à une faible réponse à l’IVIg. Neurology 2014 ; 82(10) : 879-886.
- Miura Y, et al : Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 2015 ; 138(6) : 1484-1491.
- Vural A, Doppler K, Meinl E : Autoantibodies Against the Node of Ranvier in Seropositive Chronic Inflammatory Demyelinating Polyneuropathy : Diagnostic, Pathogenic, and Therapeutic Relevance. Front Immunol 2018 ; 9 : 1029.
- Kawamura N, et al. : Anti-neurofascine antibody in patients with combined central and peripheral demyelination. Neurology 2013 ; 81(8) : 714-722.
- Devaux JJ, et al : Neurofascine-155 IgG4 dans la polyneuropathie inflammatoire démyélinisante chronique. Neurology 2016 ; 86(9) : 800-807.
- Ogata H, et al : Caractérisation de la polyneuropathie à anticorps IgG4 anti-neurofascine 155. Ann Clin Transl Neurol 2015 ; 2(10) : 960-971.
- Delmont E, et al : Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017 ; 140(7) : 1851-1858.
- Querol L, et al : Rituximab dans le CIDP résistant au traitement avec des anticorps contre les protéines paranodales. Neurol Neuroimmunol Neuroinflamm 2015 ; 2(5) : e149.
- Sommer C, et al. : Traitement des neuropathies et des neurites aiguës et chroniques à médiation immunitaire, ligne directrice S2e, 2018. Dans : Commission des lignes directrices de la Société allemande de neurologie : lignes directrices pour le diagnostic et le traitement en neurologie.
- Vedeler CA, Farbu E, Mellgren SI : Polyneuropathie inflammatoire démyélinisante chronique (PIDC). Acta Neurol Scand Suppl 2013 ; 196 : 48-51.
- Saperstein DS, et al : Spectre clinique des polyneuropathies démyélinisantes acquises chroniques. Muscle Nerve 2001 ; 24(3) : 311-324.
- Muley SA, Parry GJ : Multifocal motor neuropathy. J Clin Neurosci 2012 ; 19(9) : 1201-1209.
- Sommer C, et al : Polyneuropathies. Dtsch Arztebl Int 2018 ; 115(6) : 83-90.
- Hadden RDM, et al : Vasculitic peripheral neuropathy : Case definition and guidelines for collection, analysis, and presentation of immunisation safety data. Vaccine 2017 ; 35(11) : 1567-1578.
- Collins MP, Hadden RD : The nonsystemic vasculitic neuropathies. Nat Rev Neurol 2017 ; 13(5) : 302-316.
- Callaghan BC, et al : The Importance of Rare Subtypes in Diagnosis and Treatment of Peripheral Neuropathy : A Review. JAMA Neurol 2015 ; 72(12) : 1510-1518.
- Kadoya M, et al : IgG4 anti-neurofascine155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy : Clinical significance and diagnostic utility of a conventional assay. J Neuroimmunol 2016 ; 301 : 16-22.
- Cortese A, et al : La neurofascine-155 en tant qu’antigène putatif dans la démyélinisation centrale et périphérique combinée. Neurol Neuroimmunol Neuroinflamm 2016 ; 3(4) : e238.
- Mathey EK, et al : Autoantibody responses to nodal and paranodal antigens in chronic inflammatory neuropathies. J Neuroimmunol 2017 ; 309 : 41-46.
- Querol L, et al : Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013 ; 73(3) : 370-380.
InFo NEUROLOGIE & PSYCHIATRIE 2018 ; 16(6) : 14-18









