
L’anémie aplasique est associée à une hypocellularité de la moelle osseuse et à une pancytopénie. Chez les patients atteints d’anémie aplasique sévère ou très sévère immuno-médiée pour lesquels une greffe de moelle osseuse n’est pas envisageable, l’ajout d’un agoniste des récepteurs de la thrombopoïétine par voie orale au traitement immunosuppresseur standard s’est révélé bénéfique.
L’anémie aplasique se caractérise par un manque absolu d’érythrocytes, de leucocytes et de plaquettes en raison d’une insuffisance de la moelle osseuse. La moelle osseuse devient hypo- ou aplasique, car les cellules souches hématopoïétiques CD34-positives sont remplacées par la moelle graisseuse. Le Dr Neal S. Young, du National Heart, Lung and Blood Institute des National Institutes of Health à Bethesda (États-Unis), a expliqué lors de la réunion annuelle de l’ Association européenne d’hématologie (EHA) [1] que les mécanismes physiopathologiques varient en fonction de la cause. Les trois principaux mécanismes pathologiques sont les suivants :
- dommages chimiques ou physiques, par exemple par chimiothérapie ou radiothérapie
- défaut constitutionnel dans des gènes importants pour le maintien de l’intégrité cellulaire et la régulation immunitaire
- lésion à médiation immunitaire des cellules hématopoïétiques.
Cette dernière est la plus fréquente et est également appelée “anémie aplasique idiopathique”. On pense qu’il s’agit d’une réaction auto-immune provoquée par des lymphocytes T activés contre les cellules souches et les précurseurs sanguins. L’hémogramme montre une anémie normocytaire (MCV normal) avec une réticulocytopénie, une thrombocytopénie marquée et une leucopénie avec neutropénie. L’aspiration de la moelle osseuse ne révèle que peu ou pas de précurseurs et la biopsie de la moelle osseuse révèle une cellularité fortement réduite avec une augmentation de la moelle grasse, ce qui est caractéristique. Le tableau clinique est caractérisé par une anémie (pâleur, fatigue), une neutropénie (infection) et une thrombocytopénie (saignements) [2].
Aperçu des options de traitement
La transplantation de cellules souches hématopoïétiques est une option thérapeutique potentiellement curative . Il s’agit du traitement de choix, en particulier pour les jeunes patients ayant un donneur compatible. C’est pourquoi, dès le diagnostic, les frères et sœurs sont examinés pour déterminer leur compatibilité avec l’antigène leucocytaire humain.
Pour les patients qui ne sont pas éligibles à la transplantation ou qui n’ont pas de donneur, une alternative est le traitement immunosuppresseur par l’antithymocyte globuline de cheval (ATG) en combinaison avec la ciclosporine. Cette option de traitement est efficace chez la majorité des patients, mais pas chez tous, selon le Dr Young [1]. Environ deux tiers des patients y répondent [3]. Pendant de nombreuses années, différentes méthodes ont été testées sans succès pour améliorer les résultats du traitement immunosuppresseur standard (IST). Finalement, une percée a été réalisée : une étude de phase I/II ouverte et non randomisée a montré que l’ajout d’eltrombopag à l’ATG et à la ciclosporine chez des patients naïfs souffrant d’anémie aplasique sévère ou très sévère entraînait des taux de réponse considérables [4].
| Le critère d’évaluation principal de l’étude était une réponse hématologique complète à trois mois, définie par un taux d’hémoglobine supérieur à 10 g par dl, un nombre absolu de neutrophiles supérieur à 1000 par mm3 et un nombre de plaquettes >100 000 par mm3 chez les patients n’ayant pas reçu de transfusion. Les critères de réponse partielle étaient l’absence de transfusion (à la fois de globules rouges et de plaquettes), avec un groupe sanguin ne répondant pas aux critères d’anémie aplasique sévère, mais insuffisant pour une réponse complète. |
L’eltrombopag stimule l’hématopoïèse au niveau des cellules souches
Cet agoniste oral des récepteurs de la thrombopoïétine agit sur le contrôle de la formation des cellules souches sanguines et des plaquettes. Le médicament active la thrombopoïétine, qui contrôle la formation des plaquettes et du sang (hématopoïèse). L’étude randomisée contrôlée de phase III RACE (Randomized, Multicenter Trial Comparing Horse ATG plus Cyclosporine with or without Eltrombopag as First-Line) a comparé l’IST seul (ATG plus ciclosporine) à l’IST plus Eltrombopag comme traitement de première ligne chez des patients atteints d’anémie aplasique sévère ou très sévère [3]. Le temps médian jusqu’à la première réponse et jusqu’à la réponse complète s’est avéré plus court dans le bras d’étude avec IST plus eltrombopag (groupe expérimental, EG) par rapport au bras d’étude dans lequel IST seul a été prescrit (groupe de contrôle, KG). Ces temps de réponse plus courts expliquent pourquoi le groupe EG était auparavant indépendant des transfusions de globules rouges et de plaquettes.
- Les patients du GC ont reçu des IST consistant en ATG (40 mg par kg de poids corporel par jour pendant quatre jours consécutifs) plus de la ciclosporine orale (5 mg par kg de poids corporel par jour dès le premier jour pendant au moins 12 mois). La ciclosporine a ensuite été réduite pendant les 12 mois suivants et arrêtée après 24 mois.
- Les patients de la CE ont reçu le traitement expérimental consistant en IST plus eltrombopag (150 mg par jour à partir du jour 14). Les caractéristiques démographiques et cliniques des participants des deux groupes étaient comparables. La durée médiane de suivi a été de 24 mois.
- Le pourcentage de patients ayant complètement répondu au traitement à trois mois était de 10 % dans le groupe de référence et de 22 % dans le groupe communautaire (odds ratio groupé [OR], 3,2 ; IC à 95 % : 1,3-7,8 ; p=0,01), Tab. 1).
- Le taux de réponse global à 3 mois était plus faible dans le groupe de contrôle (31%) que dans la CE (59%) et le temps médian jusqu’à la première réponse était de 8,8 mois dans le groupe de contrôle et de 3,0 mois dans la CE. Après 12 mois, le taux de réponse complète était de 33% dans le groupe témoin et de 52% dans le groupe témoin. Le délai médian avant la première réponse était de 3,0 mois dans le bras eltrombopag et de 8,8 mois dans le bras PC.
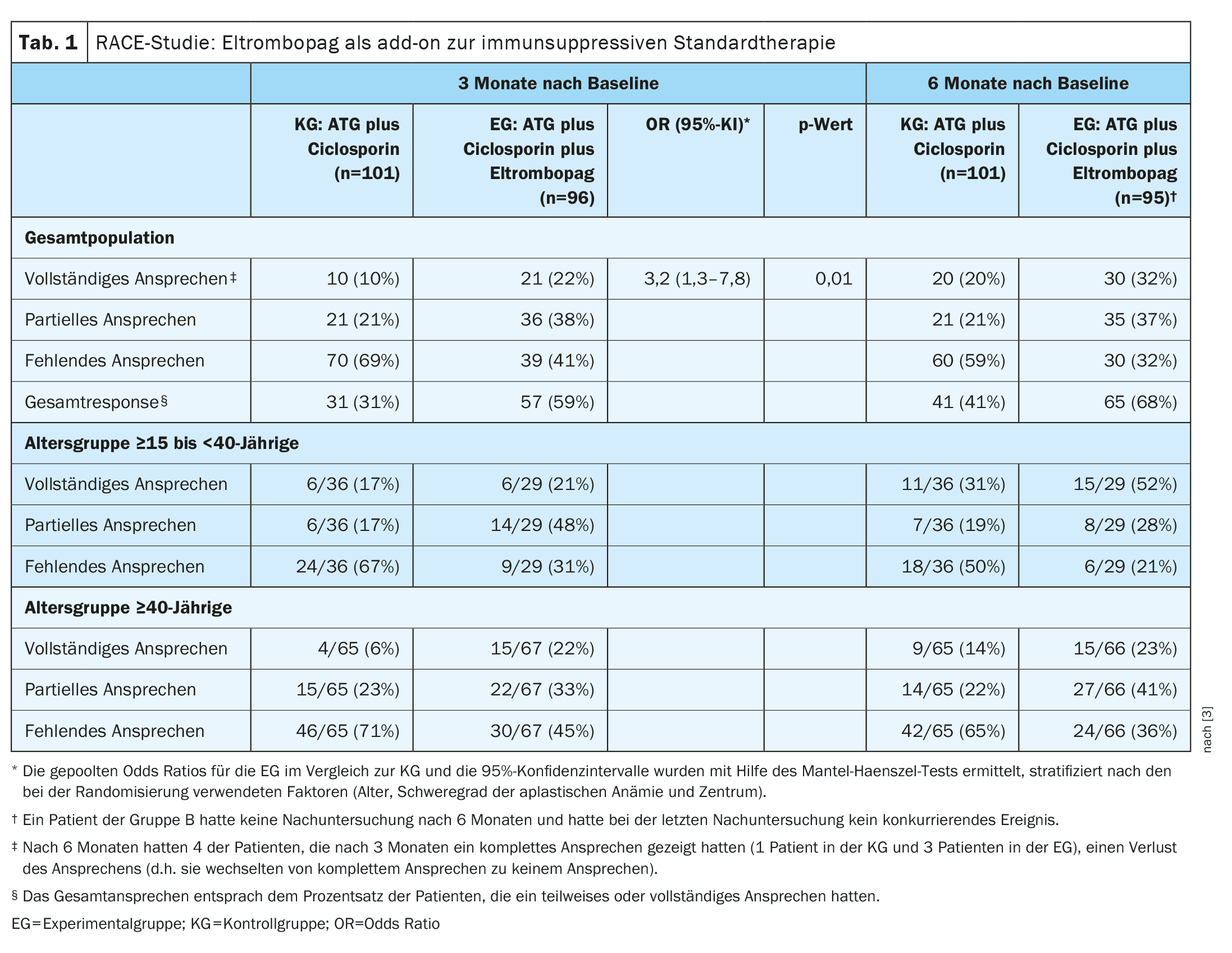
L’incidence des effets indésirables graves était comparable dans les deux groupes ; les interruptions de traitement liées au traitement ont été rares. Des mutations somatiques ont été détectées avant le début de l’étude chez 31% des patients de la CE et 29% des patients du groupe témoin. Ces pourcentages ont augmenté jusqu’à 55% dans l’étude EG et 66% dans l’étude KG à 6 mois après la ligne de base, sans effet sur la réponse hématologique ni sur les taux de survie à 2 ans (90% dans l’étude EG contre 85% dans l’étude KG).
Les greffes de moelle osseuse haplo-identiques sont également prometteuses pour élargir l’accès aux greffes de cellules souches hématopoïétiques, a mentionné le conférencier [1,5].
Congrès : EHA2024
Littérature :
- “Immune aplastic anemia : Update on Pathophysiology and New Treatments”, Dr. Neal S. Young, EHA2024, Madrid, 13.6-16.6.2024.
- Lauten M, Erlacher M, Knöfler R : Hématologie. Pédiatrie 2019 : 541-570.
- Peffault de Latour R, et al : Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. N Engl J Med 2022 ; 386(1) : 11-23.
- Townsley DM, et al : Eltrombopag ajouté à l’immunosuppression standard pour l’anémie aplasique. N Engl J Med 2017 ; 376 : 1540-1550.
- DeZern A, et al : Alternative donor BMT with post-transplant cyclophosphamide as initial therapy for acquired severe aplastic anemia [published online ahead of print, 2023 Apr 21]. Blood 2023 ; blood.2023020435. doi:10.1182/blood.2023020435.
HAUSARZT PRAXIS 2024; 19(11): 34–35 (publié le 25.11.24, ahead of print)
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(6): 26–27









