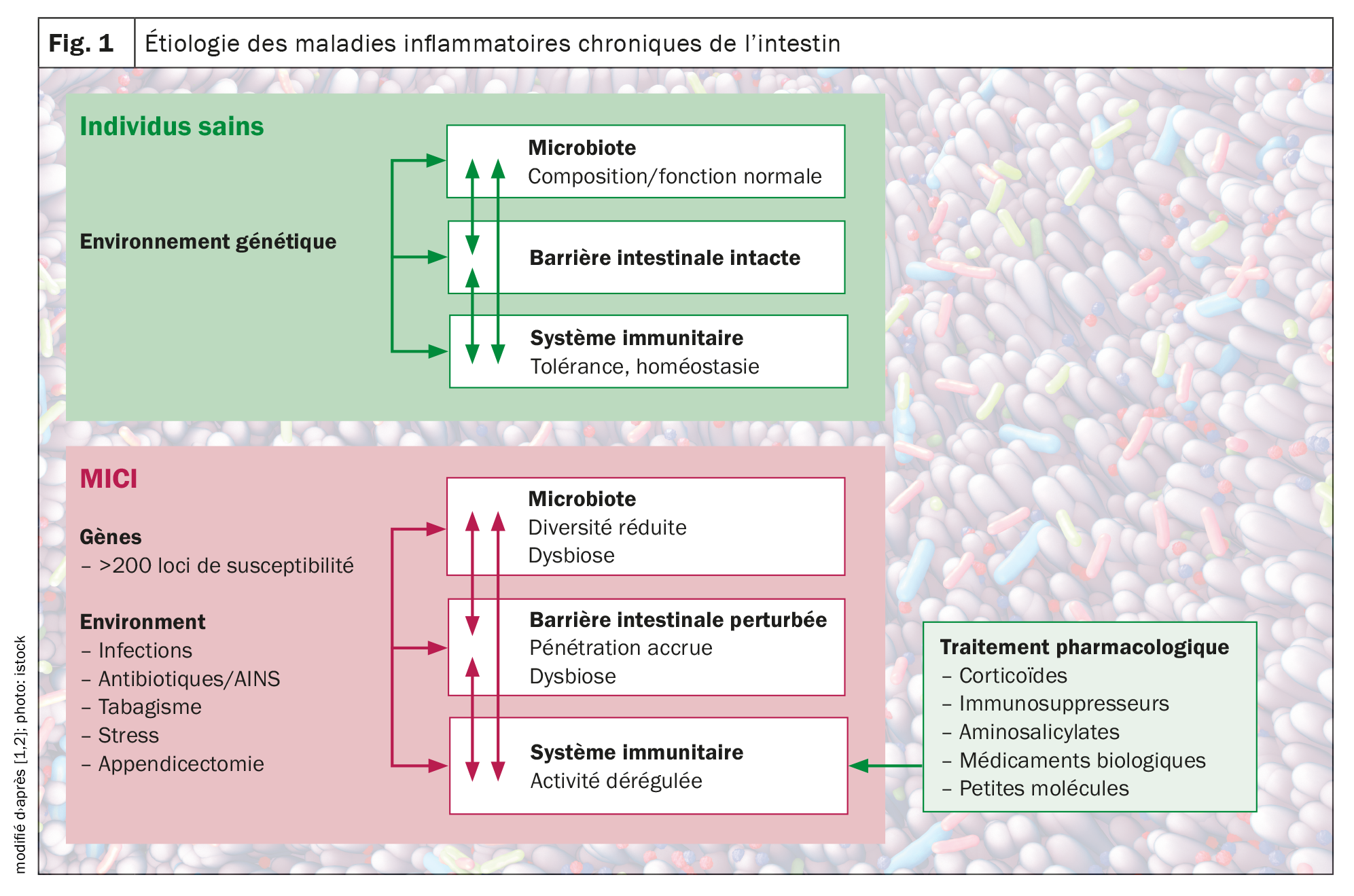
Des études ont montré qu’une altération du transport des cellules immunitaires et des cellules immunitaires pathogènes sont des facteurs déterminants de l’inflammation de la muqueuse et de la destruction des tissus dans les MICI. Une barrière intestinale défectueuse et une dysbiose microbienne entraînent une telle accumulation et une activation locale des cellules immunitaires, ce qui conduit à une boucle de cytokines pro-inflammatoires qui annule les signaux anti-inflammatoires et provoque une inflammation chronique de l’intestin.
Les maladies inflammatoires chroniques de l’intestin (MICI), telles que la maladie de Crohn (MC) et la rectocolite hémorragique (RCH), sont caractérisées par une activation incontrôlée des cellules immunitaires de l’intestin chez un individu génétiquement vulnérable. L’immunopathologie des MICI n’a pas encore été totalement élucidée à ce jour. Cependant, les composants individuels qui contribuent à la progression de ce processus inflammatoire chronique, y compris les facteurs environnementaux, les processus de migration altérés des cellules immunitaires, ainsi que les facteurs génétiques, microbiens et immunologiques, ont été continuellement étudiés. Après contact de l’organisme avec un antigène, il se produit une activation des cellules présentatrices d’antigènes (CPA) (réaction inflammatoire vs tolérogène). Les CPA peuvent produire des médiateurs tels que l’interleukine (IL-)12, ce qui entraîne l’activation, la prolifération et la différenciation des lymphocytes T avec un phénotype intestinal par une régulation à la hausse de molécules d’adhésion spécifiques. Une barrière intestinale défectueuse et une dysbiose microbienne conduisent à une accumulation et à une activation locale de cellules immunitaires, ce qui induit une boucle cytokinique pro-inflammatoire qui neutralise les signaux anti-inflammatoires et provoque une inflammation chronique de l’intestin. Des études d’association génétique ont identifié plus de 250 gènes de susceptibilité aux maladies inflammatoires de l’intestin, dévoilant des aspects fondamentaux de la biologie moléculaire de la maladie, notamment le rôle de l’autophagie et du développement et de la transmission du signal des lymphocytes T auxiliaires (Th) 17.
Outre les influences génétiques, y compris les polymorphismes génétiques de l’hôte dans un certain nombre de gènes impliqués dans la reconnaissance et la gestion des microbes, il a également été constaté que des facteurs environnementaux tels que le mode de vie, l’alimentation et les médicaments affectent l’équilibre, souvent en influençant la composition du microbiote intestinal. Il est aujourd’hui généralement reconnu que les MICI sont le résultat d’une «tempête parfaite» d’interactions entre un microbiote dysbiotique, un système immunitaire anormal et des facteurs environnementaux chez un hôte vulnérable, a expliqué le Prof. Dr Michael Scharl, directeur adjoint de la recherche et de l’enseignement à la clinique de gastroentérologie et d’hépatologie de l’Hôpital universitaire de Zurich (Fig. 1) [1,2].
Aperçu de la structure et de la fonction du système immunitaire associé à l’intestin
Cette perturbation de la barrière permet la translocation d’antigènes bactériens de l’alimentation et de certaines régions de la lumière intestinale vers la paroi intestinale, où ils rencontrent alors la plus grande concentration de cellules immunitaires du corps humain – le système immunitaire muqueux, a poursuivi le Prof. Dr Scharl. Après le contact avec l’antigène, les CPA sont activées (réaction inflammatoire vs tolérogène). Les CPA peuvent produire des médiateurs tels que l’IL-12, ce qui entraîne l’activation, la prolifération et la différenciation des lymphocytes T avec un phénotype intestinal par une régulation à la hausse de molécules d’adhésion spécifiques. Après recirculation, ces sous-groupes de lymphocytes T peuvent ensuite migrer le long de gradients chimiotactiques vers l’intestin en tant que tissu cible, où ils interagissent avec les molécules exprimées par les cellules endothéliales et initient le processus d’extravasation en plusieurs étapes de l’écotaxie («homing») intestinale. Une fois sur le site d’action, les lymphocytes T adaptent la composition de leurs molécules de surface à leur environnement, ce qui entraîne leur maintien dans les tissus ou, s’ils ne sont pas activés, leur retour dans le sang et la lymphe. Si les lymphocytes T sont activés localement par la présentation d’antigènes dans le tissu intestinal, ils peuvent causer des dommages massifs potentiels dans l’intestin enflammé [1,2].
Les réactions immunitaires dérégulées provoquent des MICI
Outre un nombre accru de lymphocytes T qui sont particulièrement répandus chez les patients atteints de MC, aussi bien les patients atteints de MC que ceux atteints de RCH présentent un nombre accru de lymphocytes Th17, qui produisent la cytokine caractéristique IL-17A. Les patients atteints de RCH présentent en outre un nombre accru de lymphocytes Th2, qui produisent par exemple de l’IL-5. Les cytokines typiques des lymphocytes Th2 sont l’IL-4 et l’IL-13. Il se produit des poussées de cellules immunitaires pro-inflammatoires activées, et ces réponses immunitaires pro-inflammatoires sont contre-régulées par des réponses immunitaires anti-inflammatoires, médiées par ex. par des lymphocytes T régulateurs (IL-10 et facteur de croissance transformant bêta [TGF-β]) ou des lymphocytes Th1. Ces cellules peuvent également être immunopathogènes et présentent les cytokines typiques suivantes: interféron gamma (IFN-γ), facteur de nécrose tumorale alpha (TNF-α).
En particulier le déséquilibre entre les cytokines pro-inflammatoires et anti-inflammatoires qui se produit dans les MICI empêche la régression de l’inflammation et conduit au contraire à la persistance de la maladie et à la destruction des tissus. Les cytokines jouent un rôle central dans la modulation du système immunitaire intestinal. Elles sont produites par les lymphocytes (en particulier les lymphocytes T de phénotype Th1 et Th2), les monocytes, les macrophages intestinaux, les granulocytes, les cellules épithéliales, les cellules endothéliales et les fibroblastes. Elles ont des fonctions pro-inflammatoires [IL-1, TNF-α, IL-12] ou anti-inflammatoires [antagoniste des récepteurs de l’IL-1 (IL-1ra), IL-10, TGF-β]. Les concentrations muqueuses et systémiques de nombreuses cytokines pro- et anti-inflammatoires sont augmentées dans les MICI. Des études d’association pangénomique ont identifié plusieurs loci de susceptibilité aux MICI contenant des gènes codant pour des cytokines et des protéines impliquées dans la signalisation des cytokines. Il a notamment été démontré que des mutations de perte de fonction dans les gènes codant pour l’IL-10 et le récepteur de l’IL-10 sont associées à des MICI de survenue très précoce [3,4].
Modifications du microbiote intestinal par les médicaments
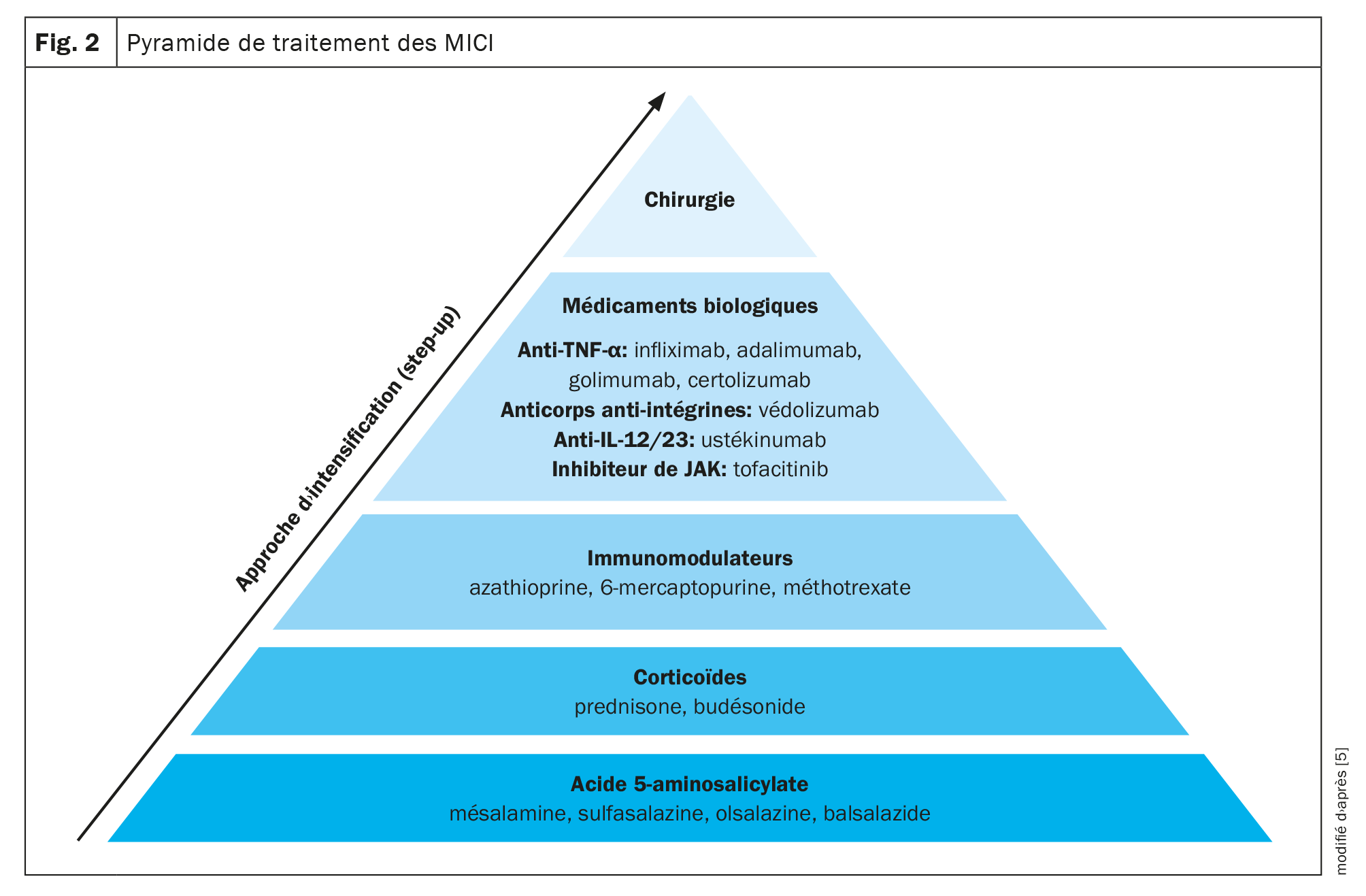
Plusieurs médicaments sont disponibles pour le traitement des MICI. Il est fréquent de recourir à une approche thérapeutique graduelle qui, en fonction de la sévérité de la MICI, passe de médicaments peu spécifiques, comme par ex. l’acide 5-aminosalicylique, à des médicaments plus puissants comme les corticoïdes, les immunomodulateurs et les agents biologiques. En plus des approches médicamenteuses, la seule autre option est la chirurgie (Fig. 2) [5].
En examinant en détail les mécanismes d’action potentiels des traitements immunomodulateurs actuellement disponibles, il apparaît qu’ils visent plusieurs cibles potentielles dans le système immunitaire muqueux, notamment les cellules immunitaires telles que les lymphocytes B, les macrophages et les lymphocytes T, ainsi que des cibles dans le domaine du transport et de la migration des lymphocytes T. Par exemple: l’ustékinumab bloque la différenciation en lymphocytes Th1 pro-inflammatoires; l’ozanimod inhibe la migration des lymphocytes T pro-inflammatoires des ganglions lymphatiques vers les vaisseaux lymphatiques efférents; le védolizumab bloque spécifiquement la migration des lymphocytes T effecteurs pro-inflammatoires des vaisseaux sanguins vers le tissu intestinal; les anti-TNF, les anti-IL-12/-23 et les inhibiteurs de Janus kinase (JAK) bloquent la fonction ou la transcription des cytokines afin de rompre le cycle inflammatoire [5].
Ustékinumab: différenciation en lymphocytes effecteurs Th1 pro-inflammatoires
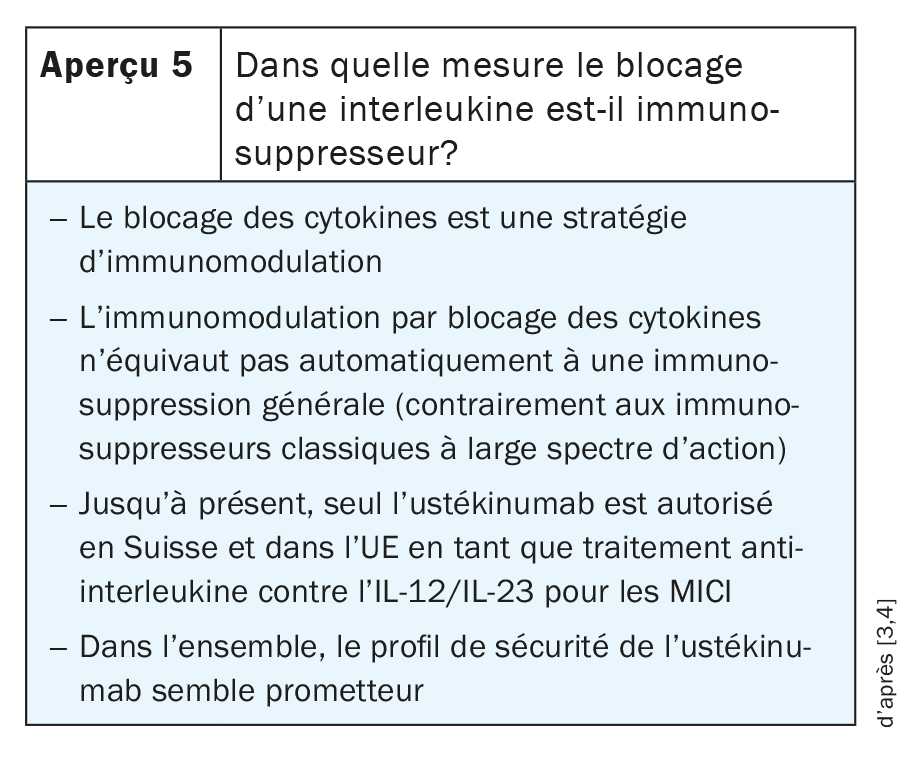
Si l’on se penche de façon approfondie sur la différenciation des lymphocytes Th dans les ganglions lymphatiques régionaux, il apparait qu’il se produit une activation de CPA qui produisent de l’IL-12. Celle-ci rencontre des lymphocytes T naïfs qui subissent une nouvelle différenciation en lymphocytes Th1. Ces lymphocytes Th1 polarisés possèdent des récepteurs d’écotaxie («homing») comme α4β7, qui leur permettent de pénétrer à nouveau dans les microbes intestinaux et d’exprimer le récepteur de l’IL-12. L’ustékinumab, qui cible l’IL-12, peut supprimer cette voie de signalisation et inhiber ainsi la polarisation des lymphocytes Th1. L’anticorps monoclonal humain se lie spécifiquement à la sous-unité p40 de l’IL-12/-23, empêchant ainsi la liaison de l’IL-12 et de l’IL-23 à leurs complexes de récepteurs de surface cellulaire, ce qui bloque les voies inflammatoires Th1 (IL-12) et Th17 (IL-23). L’ustékinumab est autorisé en Suisse aussi bien pour la MC que pour la RCH et est administré en perfusion intraveineuse pendant l’induction, à une dose d’induction de 6 mg/kg. Après une perfusion unique, on passe à un traitement d’entretien avec administration sous-cutanée de 90 mg toutes les 12 semaines/toutes les 8 semaines (Aperçu 1) [3,4].

Ozanimod: migration des lymphocytes T effecteurs pro-inflammatoires du ganglion lymphatique vers les vaisseaux lymphatiques efférents
Après l’amorçage des lymphocytes T, il se forme des cellules effectrices polarisées. Ces cellules quittent le ganglion lymphatique régional pour atteindre les vaisseaux lymphatiques efférents et la circulation sanguine. Il s’agit d’un processus actif qui génère un gradient chimiotactique, médié au moins en partie par la molécule sphingosine-1-phosphate (S1P). Cette dernière se lie au récepteur de la S1P sur les lymphocytes T, ce qui permet aux cellules de quitter le ganglion lymphatique. L’ozanimod, un agoniste des récepteurs de la S1P, intervient à ce point final du gradient et empêche les lymphocytes T de quitter le ganglion lymphatique régional. Les lymphocytes T hautement polarisés concernés ne peuvent donc plus regagner la circulation. L’agoniste des récepteurs de la S1P, qui a été préalablement étudié chez les patients atteints de sclérose en plaques, est autorisé en Suisse pour la RCH. L’ozanimod est administré par voie orale en trois phases: 0,23 mg une fois par jour du Jour 1 au Jour 4; 0,46 mg une fois par jour du Jour 5 au Jour 7; ensuite, 0,92 mg une fois par jour (Aperçu 2) [3,4].
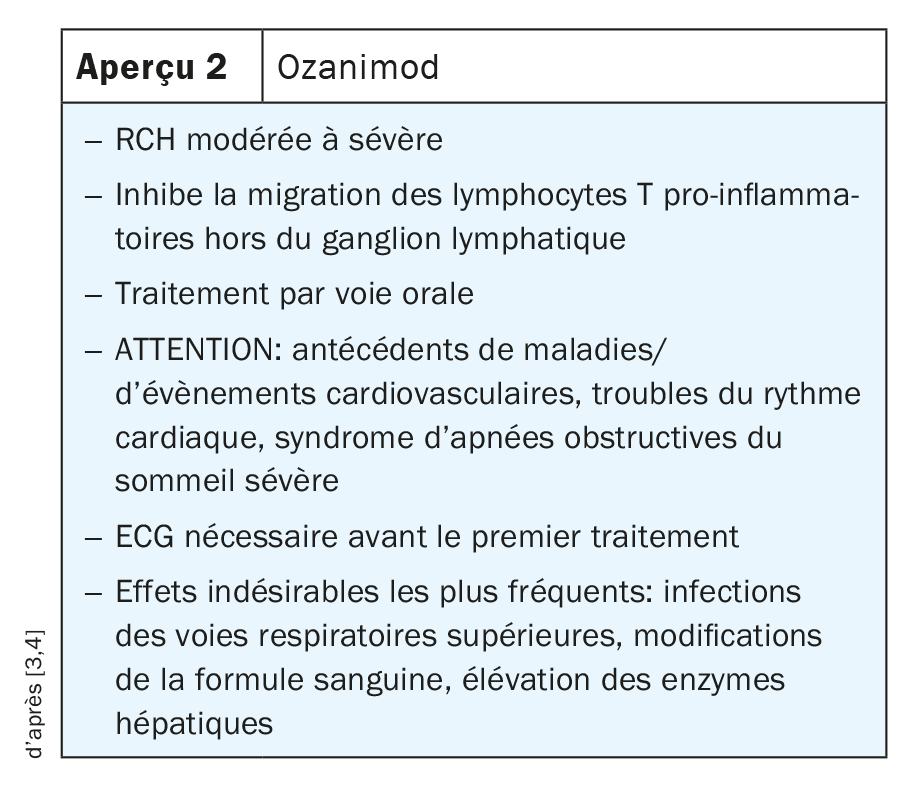
L’ozanimod est considéré comme une nouvelle option pour les patients atteints de RCH. Toutefois, étant donné qu’il s’agit d’un nouveau principe actif biologique dans le traitement des MICI, des données en vie réelle supplémentaires au-delà des études cliniques sont nécessaires pour pouvoir évaluer où l’agoniste des récepteurs de la S1P s’intègre dans le traitement des MICI, a expliqué le Prof. Dr Markus Neurath, directeur de clinique à l’Hôpital universitaire d’Erlangen. Le manque d’expérience dans la pratique clinique de routine s’illustre notamment au niveau des effets indésirables cardiovasculaires réels et de la nécessité d’un ECG, expérience nécessaire pour pouvoir finalement positionner ce médicament chez les patients atteints de RCH, a poursuivi le Prof. Dr Neurath.
Védolizumab: migration des lymphocytes T effecteurs pro-inflammatoires des vaisseaux sanguins vers le tissu intestinal
Le védolizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1). Son mécanisme d’action sélectif de l’intestin le distingue des médicaments biologiques disponibles jusqu’à présent pour le traitement des MICI, qui reposent sur une immunosuppression systémique. L’IgG1 bloque spécifiquement l’intégrine α4β7 à la surface de la sous-population de lymphocytes activés circulant dans le flux sanguin, qui sont prédisposés à un «homing» dans le tractus gastro-intestinal. Ce blocage interrompt un mécanisme physiopathologique majeur des MICI, qui permet habituellement aux lymphocytes d’adhérer à l’endothélium du tractus gastro-intestinal. Sans cette adhésion, les lymphocytes ne peuvent plus migrer de la circulation sanguine vers le tractus gastro-intestinal enflammé, ce qui réduit l’inflammation locale et crée les conditions nécessaires à un contrôle à long terme de la maladie. Le védolizumab n’interrompt pas le mécanisme de «homing» des populations de lymphocytes dans d’autres tissus, par ex. dans le système nerveux central, mais agit comme un médicament sélectif de la paroi intestinale par une immunosuppression non systémique. L’IgG1 est autorisée pour la MC et la RCH et est administrée par voie intraveineuse (300 mg aux Semaines 0, 2 et 6, puis 300 mg toutes les 8 semaines) ou sous-cutanée (300 mg aux Semaines 0 et 2, puis 108 mg toutes les 2 semaines) (Aperçu 3) [3,4,6].

Effets pro-inflammatoires pléiotropes du TNF
Le TNF est un médiateur décisif dans le contrôle des processus inflammatoires dans l’intestin et il est utilisé depuis plus de 20 ans dans la routine clinique. Le TNF et ses récepteurs jouent un rôle déterminant dans la pathogenèse des MICI. Ainsi, des concentrations élevées de la forme soluble du récepteur 1 du TNF (TNFR1) et du récepteur 2 du TNF (TNFR2) ont été constatées à la fois chez les patients atteints de MC et chez ceux atteints de RCH, et leur expression était corrélée à l’activité de la maladie.
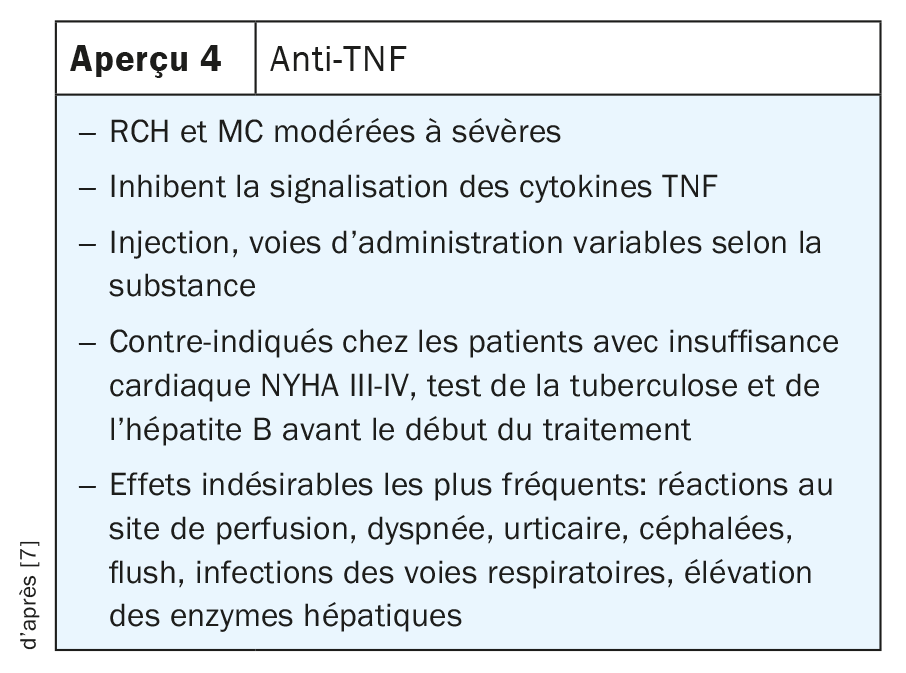
Les anticorps anti-TNF bloquent à la fois la forme précurseur transmembranaire (mTNF) et la forme soluble (sTNF), réduisant ainsi le milieu pro-inflammatoire dans l’intestin en bloquant l’interaction entre le TNF et son récepteur, ce qui bloque différents types de cellules immunitaires pro-inflammatoires. De plus, le TNF provoque la mort des cellules épithéliales. Les anticorps anti-TNF possèdent donc plusieurs mécanismes d’action qui peuvent être utilisés dans la pratique clinique chez les patients atteints de MICI, tant pour la MC que pour la RCH. Parmi les anticorps anti-TNF éprouvés pour la pratique clinique de routine figurent l’infliximab, l’adalimumab, le golimumab et le certolizumab pégol, dont l’utilisation diffère. Certains d’entre eux sont disponibles pour le traitement intraveineux, d’autres sont disponibles à la fois pour le traitement sous-cutané et intraveineux, et d’autres encore ne sont disponibles que pour l’administration sous-cutanée (Aperçu 4) [7].
Selon le Prof. Dr Neurath, les différents anticorps anti-TNF ont fait leurs preuves dans la pratique clinique et sont encore utilisés aujourd’hui de manière ciblée. Le choix entre la voie intraveineuse et la voie sous-cutanée dépend un peu de l’environnement clinique. En cas d’activité clinique élevée ou si le patient est hospitalisé, l’administration intraveineuse est certainement un choix judicieux pour l’administration d’anticorps anti-TNF, en particulier chez les patients présentant une activité très élevée qui perdent beaucoup d’anticorps dans les selles. Il n’est pas absolument indispensable de mesurer les concentrations résiduelles ou de vérifier le statut des anticorps, a ajouté le Prof. Dr Neurath. En général, cela se fait uniquement chez les patients qui ne répondent pas, qui présentent une perte d’efficacité secondaire ou qui n’obtiennent pas la réponse clinique primaire souhaitée. Dans ce cas, il y a plusieurs options, a poursuivi le Prof. Dr Neurath: soit passer à un autre principe actif, soit ajouter un immunosuppresseur comme l’azathioprine pour supprimer les réponses des lymphocytes B et les anticorps anti-médicament. L’étude SONIC a déjà montré qu’un traitement combiné, associant par exemple l’azathioprine et l’infliximab, est supérieur à une monothérapie avec un seul des deux médicaments. Un traitement combiné devrait toutefois dépendre de l’activité clinique. Alternativement, le patient peut faire l’objet d’une surveillance clinique, par exemple par des échographies, le dosage de la protéine C réactive (CRP) ou la détermination de l’activité clinique, afin de déterminer si le patient est en rémission clinique. Si des problèmes surviennent, il est alors possible soit d’intensifier le traitement, par ex. en raccourcissant l’intervalle entre les perfusions et en augmentant la dose du principe actif, soit de passer à une autre classe de principes actifs biologiques.
Activité des lymphocytes Th17 effecteurs
pro-inflammatoires
L’ustékinumab est un anticorps qui bloque non seulement l’IL-12, mais aussi l’IL-23. L’IL-23 entraîne l’activation et le maintien des fonctions effectrices des lymphocytes Th17 pro-inflammatoires dans le tissu intestinal. L’ustékinumab réduit l’activité des lymphocytes Th17 pro-inflammatoires dans le tissu intestinal en bloquant l’interaction Il-23/Il-23R. Sur la base de ces connaissances, des efforts sont actuellement déployés pour développer des antagonistes sélectifs de l’IL-23. Ces derniers ne devraient pas cibler la sous-unité p40, comme c’est le cas avec l’ustékinumab, mais la sous-unité p19, qui est unique à l’IL-23 et n’est pas présente dans l’IL-12. Certains principes actifs sont déjà en phase d’essai clinique et seront tôt ou tard utilisés dans la pratique clinique, comme le risankizumab, le mirikizumab, le guselkumab et le brazikumab. En outre, certaines données préliminaires indiquent que les inhibiteurs de p19 pourraient également être efficaces lorsque les inhibiteurs de p40 n’ont pas fonctionné auparavant (Aperçu 5) [3,4].
Transduction du signal des cytokines via les voies de signalisation JAK/STAT
La voie de signalisation JAK et transducteur de signal et activateur de la transcription (JAK-STAT) joue un rôle majeur dans la transmission des signaux des récepteurs membranaires cellulaires vers le noyau cellulaire. De nombreuses cytokines pro-inflammatoires induisent la transcription de gènes effecteurs dans la cellule cible en activant des voies de signalisation JAK/STAT spécifiques. La famille des JAK humaines se compose de quatre membres: JAK1, JAK2, JAK3 et TYK2. Le tofacitinib, un inhibiteur qui cible principalement JAK1 et JAK3, et dans une moindre mesure JAK2, réduit la transcription des gènes de signalisation et des gènes effecteurs pro-inflammatoires en bloquant l’activité kinase des JAK. Le tofacitinib est une petite molécule («small molecule») qui agit simultanément sur plusieurs cytokines et qui peut être administrée par voie orale. Jusqu’à présent, l’inhibiteur est toutefois uniquement autorisé pour la RCH, mais pas pour la MC (Aperçu 6) [8–10].
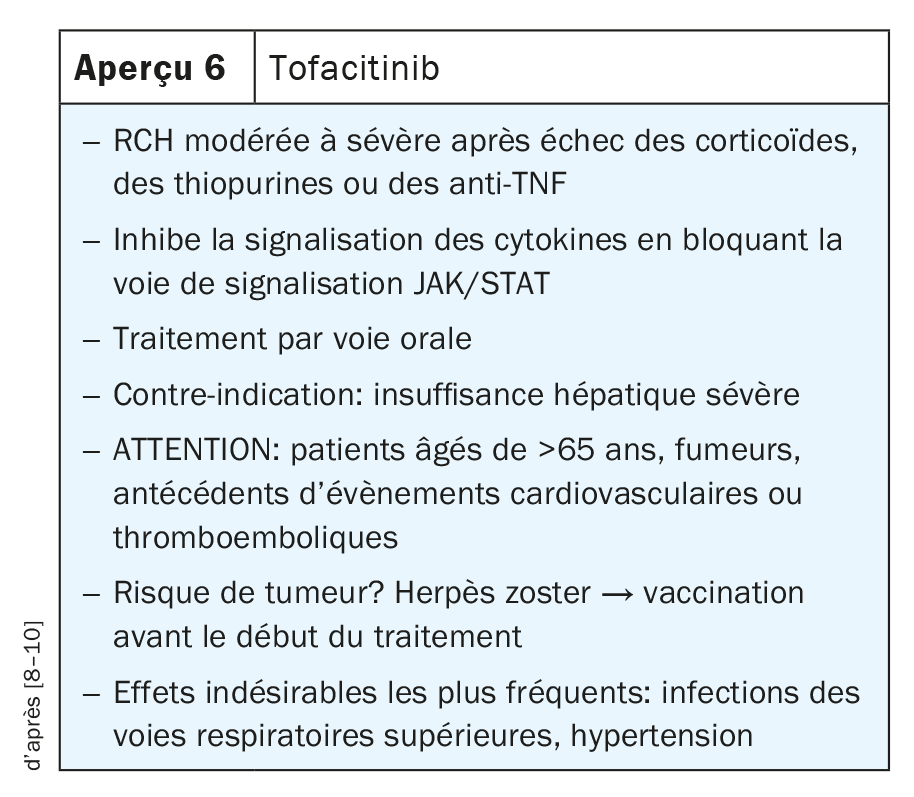
En raison de son activité immunomodulatrice et du risque d’évènements cardiovasculaires et thromboemboliques, des restrictions d’utilisation ont été imposées depuis l’autorisation de mise sur le marché du tofacitinib, ce qui n’en fait pas un médicament de premier choix, selon le Prof. Dr Neurath. En particulier chez les patients âgés, chez ceux présentant une maladie cardiovasculaire et chez ceux ayant un risque potentiellement accru d’évènements thromboemboliques, une évaluation minutieuse s’impose avant l’instauration du traitement.
En Suisse, seul le tofacitinib est actuellement disponible pour l’inhibition de JAK. Cependant, compte tenu de la diversité des études, les inhibiteurs de JAK sont de plus en plus souvent autorisés ou font l’objet d’études cliniques, de sorte que tôt ou tard, toute une série d’inhibiteurs de JAK seront disponibles pour la pratique clinique. Dans ce contexte, des modifications infimes de l’affinité des molécules feront une grande différence clinique. D’après le Prof. Dr Neurath, il pourrait par exemple y avoir des différences pertinentes entre les divers inhibiteurs de JAK1, mais cela doit encore être étudié. La pathogenèse montre clairement que ces principes actifs interviennent dans l’activation des cellules immunitaires. Il sera intéressant de comparer leur efficacité, mais aussi et surtout leur profil de sécurité, a ajouté le Prof. Dr Neurath. En effet, en particulier la sécurité joue un rôle décisif pour la routine clinique, mais aussi pour les patients.

Options futures et traitements combinés pour les MICI
Selon le Prof. Dr Neurath, les premières données d’une étude associant des inhibiteurs de p19 et des anticorps anti-TNF sont disponibles et d’autres études sont en cours [11]. Il s’agissait essentiellement d’un traitement combiné visant à induire une rémission, et non d’un traitement combiné à vie. Cela pourrait toutefois être une option pour les patients difficiles à traiter. Le védolizumab, en particulier, semble être un partenaire intéressant pour les traitements combinés, car il possède un mécanisme d’action moléculaire totalement différent de celui des autres principes actifs, de sorte que l’on peut facilement postuler qu’il pourrait y avoir des synergies, a précisé le Prof. Dr Neurath. Qui plus est, c’est un principe actif très sûr, qui présente un bon profil de sécurité, si bien qu’il constitue un élément de base idéal pour toute approche de traitement combiné. En revanche, le Prof. Dr Neurath ne voit pas de grand potentiel dans l’association d’inhibiteurs de JAK avec des principes actifs biologiques.
Take-Home-Messages
- Les traitements immunomodulateurs visent différentes cibles potentielles dans le système immunitaire muqueux.
- L’ustékinumab bloque la différenciation en lymphocytes Th1 (via l’IL-12), ainsi que la cytokine IL-23.
- L’ozanimod inhibe la migration des lymphocytes T pro-inflammatoires du ganglion lymphatique vers les vaisseaux lymphatiques efférents.
- Le védolizumab bloque spécifiquement la migration des lymphocytes T effecteurs pro-inflammatoires des vaisseaux sanguins vers le tissu intestinal.
- Les anti-TNF, les anti-IL-12/-23 et les inhibiteurs de JAK bloquent la fonction des cytokines afin de stopper l’inflammation.

Références:
- Neurath MF: Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol 2019; doi: 10.1038/s41590-019-0415-0.
- de Lange KM, et al.: Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017; doi: 10.1038/ng.3760.
- Neurath MF: Cytokines in inflammatory bowel disease. Nat Rev Immunol 2014; doi: 10.1038/nri3661.
- Neurath MF: Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 2017; doi: 10.1038/nrgastro.2016.208.
- Wu N, et al.: Inflammatory bowel disease and the gut microbiota. Proc Nutr Soc 2021; doi: 10.1017/S002966512100197X.
- Denucci CC, et al.: Integrin function in T-cell homing to lymphoid and nonlymphoid sites: getting there and staying there. Crit Rev Immunol 2009; doi: 10.1615/critrevimmunol.v29.i2.10.
- Billmeier U, et al.: Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol 2016; doi: 10.3748/wjg.v22.i42.9300.
- Vetter M, Neurath M: Emerging oral targeted therapies in inflammatory bowel diseases: opportunities and challenges. Therap Adv Gastroenterol 2017; doi: 10.1177/1756283X17727388.
- Seif F, et al.: The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal 2017; doi: 10.1186/s12964-017-0177-y.
- Danese S, et al.: JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut 2019; doi: 10.1136/gutjnl-2019-318448.
- Feagan BG, et al.: Guselkumab plus golimumab combination therapy versus guselkumab or golimumab monotherapy in patients with ulcerative colitis (VEGA): a randomised, double-blind, controlled, phase 2, proof-of-concept trial. Published: February 01, 2023DOI:https://doi.org/10.1016/S2468-1253(22)00427-7.
HAUSARZT PRAXIS 2023; 18(7): 4–9