
En plus de la prise en charge standard de toutes les formes de SLA, des progrès significatifs ont été réalisés, en particulier pour les formes monogéniques de SLA, grâce au développement de méthodes thérapeutiques spécifiques basées sur les gènes, notamment actuellement l’ASO. Ainsi, un diagnostic génétique de base, au moins pour les gènes les plus courants (SOD1, C9orf72, FUS, TARDP), est recommandé pour tous les patients atteints de SLA au moment du diagnostic.
Vous pouvez passer le test de FMC sur notre plateforme d’apprentissage après avoir consulté le matériel recommandé. Pour ce faire, veuillez cliquer sur le bouton suivant :
Les cas de sclérose latérale amyotrophique (SLA) ont été décrits pour la première fois par Jean Martin Charcot en 1873 [1]. Charcot avait déjà apporté la preuve neuropathologique d’une dégénérescence du système moteur sous-jacent. On sait maintenant que la sclérose latérale amyotrophique est une neurodégénérescence multisystémique avec de nombreuses manifestations extra-motrices, malgré la dégénérescence des premier et deuxième motoneurones et du tractus cortico-spinal. Nous avons acquis de plus en plus de connaissances sur les facteurs génétiques sous-jacents, en particulier au cours de la dernière décennie, ce qui a conduit aux premières conséquences thérapeutiques immédiates.
Épidémiologie
Les données du registre des patients le mieux tenu en Souabe permettent d’estimer qu’il y a environ 8000 à 9000 personnes atteintes de SLA dans l’ensemble de l’Allemagne [2]. Avec un âge moyen de la maladie de 70 à 75 ans et une légère prédominance masculine, on estime l’incidence à environ 3/100 000 patients. La prévalence sur la vie entière, qui est la mesure statistique la plus claire de la probabilité de développer une SLA, est de 1/400. Ces mesures épidémiologiques sont très proches de celles d’autres pays européens. Dans d’autres parties du monde, comme l’Asie, les données épidémiologiques sont différentes [3,4].
Symptômes cliniques
Les principaux symptômes de la SLA sont des parésies atrophiques focales et progressives avec des crampes et des fasciculations musculaires fréquentes, signe d’une atteinte croissante des seconds motoneurones au niveau de la colonne vertébrale ou du tronc cérébral [5]. L’atteinte des premiers motoneurones du cortex moteur primaire et du tractus corticospinal peut précéder ou accompagner l’évolution de la maladie, avec une augmentation et un saut des réflexes d’extension musculaire et la mise en évidence de réflexes pathologiques ou une augmentation du tonus musculaire dans le sens d’une spasticité.
Des déficits cognitifs et des troubles du comportement cliniquement et quotidiennement significatifs, dans le sens d’un syndrome de démence frontotemporale, sont observés en tant que symptomatologie extra-motrice chez environ 5% des personnes atteintes de SLA [6,7]. Des troubles associés du système nerveux autonome ont été décrits de plus en plus fréquemment ces dernières années [8]. En outre, des douleurs d’origines diverses sont également importantes au cours de l’évolution de la maladie [9].
Procédures de diagnostic et établissement du diagnostic
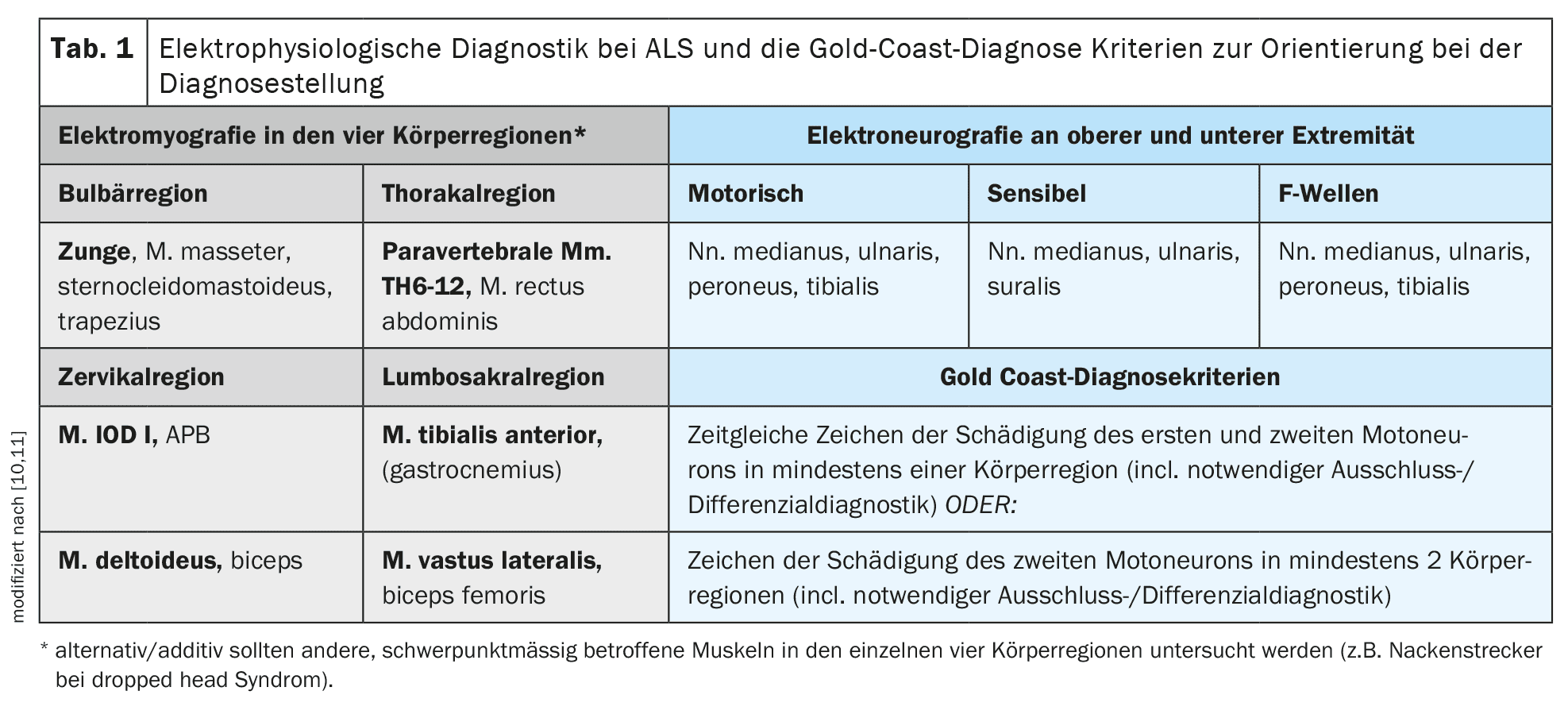
Le diagnostic est avant tout clinique, après avoir exclu soigneusement et systématiquement les autres diagnostics différentiels. Les critères de Gold Coast [10] fournissent une bonne orientation pour le diagnostic clinique. Le principal outil de diagnostic est l’électromyographie, qui permet de mettre en évidence une dénervation active ou des potentiels de fasciculation polytopériques en présence d’une lésion neurogène chronique. L’électromyographie doit examiner différents muscles des quatre régions du corps (nerfs crâniens/bulbaire – cervical – thoracique – lombosacré) [11]. En complément de l’électromyographie, une échographie musculaire peut être réalisée afin de déterminer la présence de fasciculations musculaires polytopériques et d’évaluer la trophicité musculaire et la structure interne. La sonographie musculaire peut être particulièrement utile pour détecter les fasciculations musculaires dans les muscles plus grands ou plus profonds, tels que les parties du muscle quadriceps fémoral ou les muscles de la base de la langue, et peut constituer un complément important à l’électromyographie. L’électroneurographie des nerfs moteurs et sensitifs ainsi que le diagnostic des ondes F au niveau des membres supérieurs et inférieurs sont particulièrement nécessaires pour exclure une neuropathie démyélinisante primaire et surtout des blocs de conduction (CIDP ou neuropathie motrice multifocale/MMN).
Pour objectiver et, le cas échéant, quantifier la participation des premiers motoneurones, on dispose de la stimulation magnétique transcrânienne pour la dérivation des potentiels évoqués moteurs. Cette méthode d’examen permet d’étudier la conduction motrice centrale des axones épais et myélinisés du tractus corticospinal depuis le cortex moteur en tant que lieu de stimulation tout au long de la moelle épinière [12]. Il est utile d’examiner un muscle distal du membre supérieur et un muscle distal du membre inférieur. Le tableau 1 présente un aperçu détaillé du diagnostic électrophysiologique proposé pour la SLA et des critères de diagnostic de Gold Coast .
Les chaînes légères du neurofilament (NfL) constituent un biomarqueur important [13]. Elles peuvent être facilement déterminées dans le sérum et le LCR. Il est important que la détermination soit effectuée avec une méthode de détection suffisamment sensible dans un laboratoire établi avec des valeurs limites adaptées à l’âge. Il convient de souligner que si la NfL peut constituer un élément supplémentaire important pour l’établissement du diagnostic, une NfL dans la norme ne permet pas d’exclure le diagnostic de SLA et des valeurs élevées de NfL peuvent également être observées dans d’autres diagnostics différentiels de la SLA (CIDP, neuropathie vasculaire, neuropathie associée à l’amylose, MMN) [14]. L’importance des NfL en tant que marqueur pronostique de l’évolution de la maladie semble être encore plus grande que dans le diagnostic différentiel, bien que, là encore, aucune conclusion fiable ne puisse être tirée de la valeur des NfL pour chaque patient.
Si le diagnostic génétique était encore considéré comme facultatif dans le guide S1 disponible depuis 2021, les développements des trois dernières années ont modifié cette situation selon les auteurs [5]. Compte tenu des développements thérapeutiques dans ce domaine, chaque patient atteint de SLA devrait obligatoirement subir un test génétique ciblé au moins sur la présence d’une altération du gène de la superoxyde dismutase 1 Cu/Zn(SOD 1) et les jeunes patients SLA de moins de 40 ans sur la présence d’une mutation pathogène du gène FUS [15,16]. En option, un diagnostic génétique plus poussé devrait être réalisé avec l’étude d’autres gènes tels que C9orf72, TARDP, TBK1, etc. [17]. Les sections suivantes sur l’étiologie et la génétique, et en particulier sur le traitement, aborderont ce sujet de manière plus détaillée. Le tableau 1 donne un aperçu des méthodes de diagnostic présentées et des critères de diagnostic de la Côte d’Or recommandés dans la pratique clinique .
En ce qui concerne les autres examens cliniques pertinents pour un diagnostic différentiel et d’exclusion complet en fonction de la présentation clinique initiale (par ex. imagerie IRM, diagnostic ORL, diagnostic de laboratoire) ainsi que les diagnostics pertinents au cours de l’évolution pour l’évaluation du pronostic (par ex. test de la fonction pulmonaire/diaphragmatique, diagnostic de la déglutition par FEES, psychométrie par ECAS), nous souhaitons renvoyer explicitement à la présentation très détaillée et claire de la ligne directrice S1 à ce sujet [5].
Spectre phénotypique et modèle des parésies musculaires dans le contexte de la neuroanatomie et de la physiopathologie de la SLA
Les découvertes scientifiques des deux dernières décennies, en particulier les études neuroanatomiques et neuropathologiques, ont radicalement changé la vision de la SLA. Ainsi, de nos jours, la SLA n’est plus considérée comme une dégénérescence du système purement moteur, mais comme une dégénérescence multisystémique [18,19].
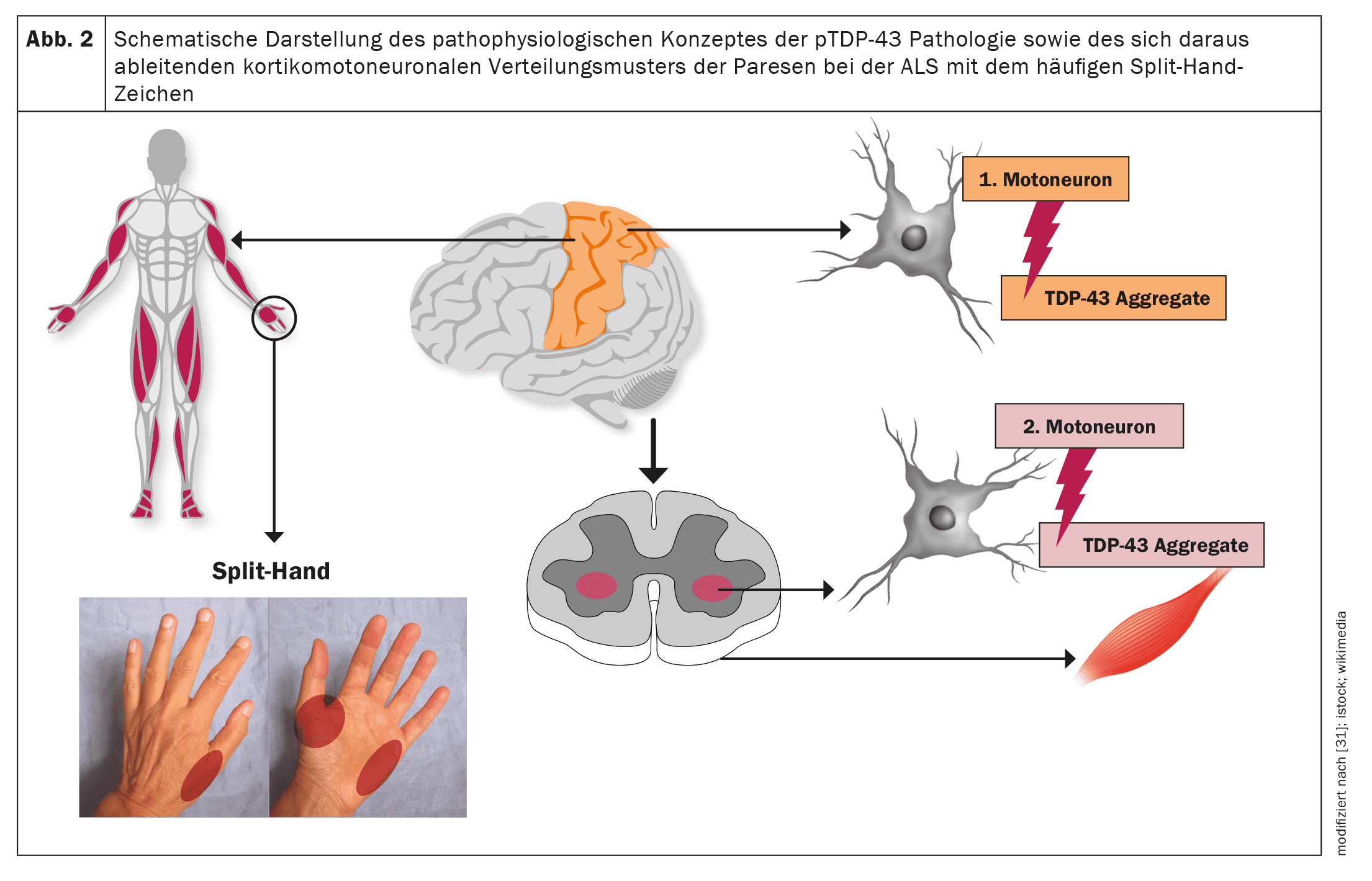
La SLA, comme d’autres maladies neurodégénératives telles que la maladie d’Alzheimer ou la maladie de Parkinson, est une protéinopathie, c’est-à-dire que des agrégats de protéines pathologiques dans les neurones moteurs, en tant que corrélat neuropathologique, conduisent à un dysfonctionnement des neurones moteurs concernés et finalement à l’apoptose et donc à la perte de neurones moteurs. Dans le cas de la SLA, il s’agit dans plus de 95% des cas d’agrégats de protéines TDP-43 [20]. Seuls les variantspathologiques SOD1 ou FUSentraînent une neurodégénérescence indépendante du TDP-43 [21]. Les études neuropathologiques ont permis d’établir une propagation cérébrale progressive, par étapes, de la pathologie du TDP-43 dans la SLA, comparable à la pathologie de l’α-synucléine dans la maladie de Parkinson et à la pathologie tau dans la maladie d’Alzheimer [22]. Ces résultats, ainsi que le schéma corticomotoneuronal des parésies avec le signe de la main divisée (atrophie asymétrique des C8/T1 ou des cubital) comme signe clinique très fréquent dans la SLA implique une origine des modifications neuropathologiques dans la région du cortex moteur primaire et une propagation progressive de la pathologie TDP-43 de là via un transport axonal vers les seconds motoneurones, c’est-à-dire les noyaux des nerfs moteurs cérébraux dans le tronc cérébral et les cellules de la corne antérieure de la moelle épinière [23–26].
Il semble qu’il y ait une propagation de type prion des modifications neuropathologiques, ce qui expliquerait le début focal des manifestations motrices ainsi qu’une extension progressive aux myotomes et aux régions corporelles voisines [27,28]. En conséquence, selon la manifestation initiale de la neuropathologie, le spectre phénotypique de la SLA – concernant uniquement les symptômes moteurs – n’est pas uniforme mais très variable [29]. Ainsi, des phénotypes moteurs distincts sur le plan topographique peuvent apparaître. De plus, le phénotype est influencé par la vitesse généralement différente de dégénérescence du premier et du second motoneurone et par les symptômes cliniques associés. En l’absence d’une compréhension précise des mécanismes moléculaires, le rapport entre les agrégats de protéines TDP-43 insolubles, qui ne peuvent plus être transportés axonalement et qui s’accumulent donc localement, et les oligomères TDP-43 solubles, précurseurs des agrégats TDP-43, semble être déterminant. En effet, il est évident que ces précurseurs solubles sont ensuite transportés par voie axonale des premiers motoneurones vers les seconds, pour s’y déposer obligatoirement sous forme d’agrégats de protéines et conduire à la destruction de ces cellules [23].
Il n’existe pas de classification uniforme des phénotypes cliniques qui tienne compte de la proportion ou de l’importance des lésions du premier et du deuxième motoneurone, du schéma clinique ou de l’importance des parésies atrophiques et de leur extension.
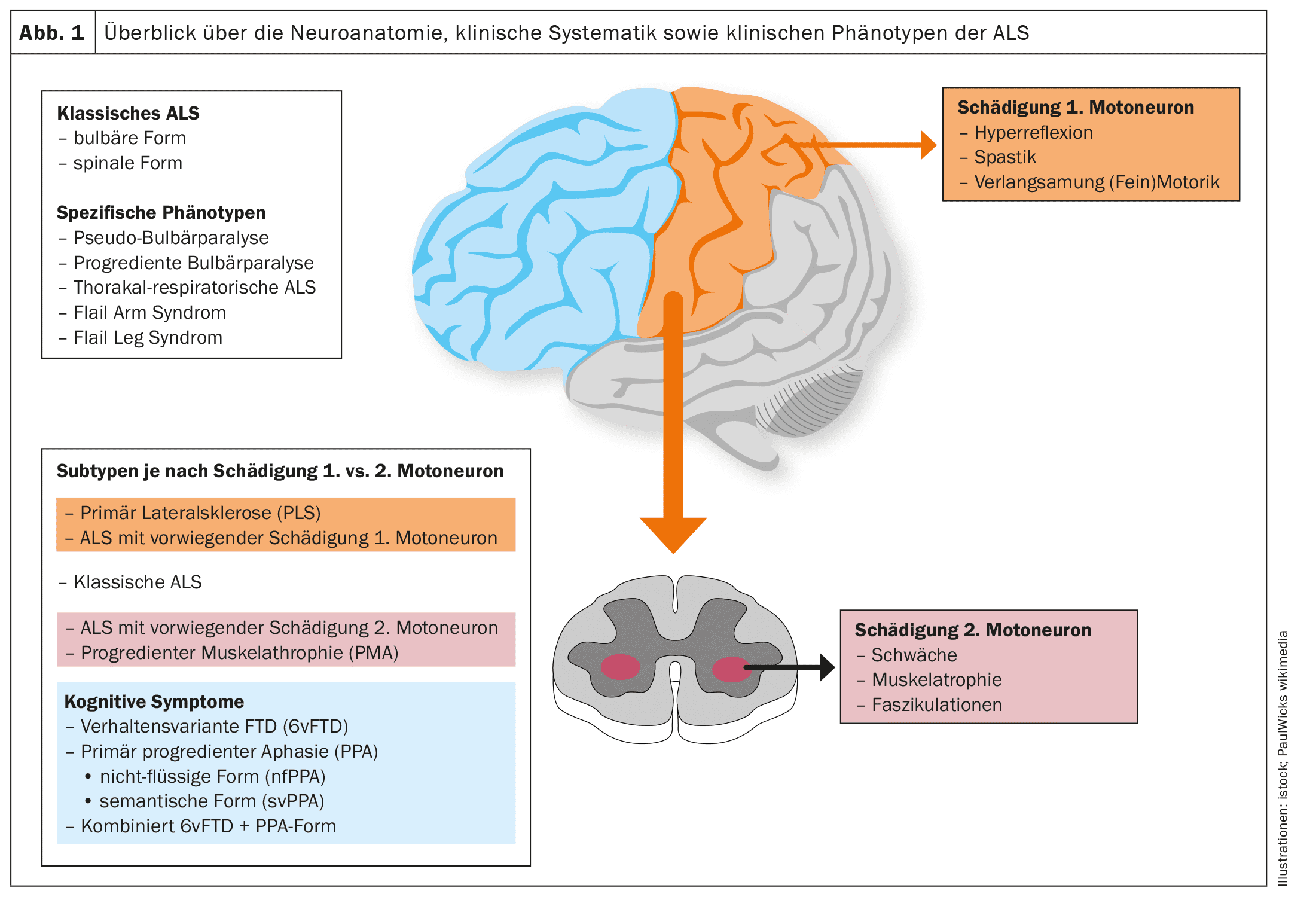
Les figures 1 et 2 offrent un aperçu schématique de la neuroanatomie et de la physiopathologie de la maladie, ainsi que des phénotypes cliniques et de leur systématique.
Étiologie et génétique
L’étiologie de la SLA reste mal comprise. Dans la forme sporadique de la maladie, qui représente la grande majorité des cas, on pense que l’origine est multifactorielle, avec l’interaction de différents facteurs environnementaux externes défavorables, de facteurs métaboliques épigénétiques et d’une éventuelle susceptibilité génétique supplémentaire. Les mécanismes métaboliques intracellulaires sous-jacents sont des altérations du transport des protéines nucléaires-cytosoliques, du métabolisme de l’ARN, de la fonction cellulaire oxydative mitochondriale, de l’excitabilité glutamatergique, du transport axonal des protéines et de l’autophagie cellulaire, et les mécanismes extracellulaires additifs sont des dysfonctionnements des atrocytes et des oligodendrocytes et des processus inflammatoires [31]. Des connaissances essentielles sur les mécanismes étiologiques possibles de la SLA sporadique ont été acquises principalement sur des formes génétiques avec des processus physiopathologiques distincts.
En 1993, une cause génétique de la SLA a été décrite pour la première fois avec des mutations pathogènes dans la superoxyde dismutase 1 Cu/Zn (SOD1) [32]. Aujourd’hui, un grand nombre de mutations génétiques sont connues comme étant l’étiologie sous-jacente de la SLA. Dans le cas de la SLA apparemment sporadique sans antécédents familiaux particuliers en Allemagne, une cause monogénique peut être identifiée dans un peu plus de 10% des cas [17]. Les causes génétiques les plus fréquentes en Allemagne sont des expansions pathologiques de répétitions C9orf72 et des mutations dans les gènes SOD1, TARDP et FUS [17].
Un diagnostic génétique au moment du diagnostic devrait donc impérativement être proposé à tout patient atteint de SLA, car il peut désormais avoir des conséquences thérapeutiques immédiates, que nous aborderons en détail dans le paragraphe suivant.
Traitement et pronostic
Le riluzole est la seule substance pharmacologique dont l’effet positif sur la progression de la SLA a été prouvé et qui est autorisée en Allemagne depuis 1996. Le mécanisme d’action décisif postulé est une réduction de la libération de glutamate et donc une réduction de l’excitotoxicité. Cette hypothèse a été renforcée par la mise en évidence d’une dégénérescence primaire des voies cortico-fugales glutamatergiques [18]. Une dose quotidienne de 100 mg de riluzole présente le meilleur profil effet/effet indésirable. Les analyses rétrospectives de dix registres cliniques de la SLA indiquent que le riluzole prolonge la durée de survie de 19 mois en moyenne et qu’il est clairement efficace à un stade avancé de la maladie [33–35]. Le riluzole est généralement bien toléré, mais les effets secondaires connus incluent une augmentation des transaminases, qui doivent donc être surveillées régulièrement, ainsi que des troubles gastro-intestinaux. Il existe désormais d’autres formes galéniques pour les patients atteints de SLA et présentant des troubles de la déglutition, comme le jus ou les comprimés à avaler.
Outre le traitement pharmacologique par le riluzole, la prévention d’un état métabolique catabolique avec perte de poids consécutive est considérée comme un avantage pronostique supplémentaire. Plus l’indice de masse corporelle (IMC) est élevé, plus le pronostic est favorable [36].
Actuellement, les recommandations aux patients atteints de SLA sont de maintenir un poids stable et d’éviter la perte de poids. Dans ce contexte, la mise en place d’une sonde PEG joue un rôle important en cas de troubles progressifs de la déglutition, avec un allongement de la durée de survie grâce à cette mesure [37]. Des études approfondies sont actuellement en cours pour déterminer si et dans quelle mesure des approches thérapeutiques anticataboliques spécifiques, telles que des interventions nutritionnelles ciblées riches en graisses et à haute teneur calorique ou une intervention nutritionnelle cétogène, peuvent améliorer le pronostic. Dans ce contexte, l’étude LIPCAL-ALS 2 menée à l’échelle allemande par les collègues d’Ulm en tant qu’Investigator Initiated Trial (IIT), qui devrait débuter entre 2024 et 2025, est d’une grande importance.
En cas d’insuffisance ventilatoire, la ventilation non invasive (VNI) et invasive par trachéotomie [38,39] est une autre mesure essentielle pour prolonger la survie dans la SLA. Cela est également plausible, car une hypoventilation alvéolaire avec hypercapnie consécutive sur fond d’atteinte diaphragmatique est typiquement un facteur essentiel de décès des patients atteints de SLA après trois à cinq ans d’évolution de la maladie.
Le développement d’oligonucléotides antisens (ASO) pour des formes spécifiques de SLA génétiquement induites constitue une étape importante. Il s’agit notamment de l’ASO Tofersen administré par voie intrathécale, qui se lie à l’ ARNm de la SOD1chez les personnes atteintes d’une variantepathogène de la SOD1(environ 1 à 2% de tous les cas de SLA) et empêche ainsi l’expression cytotoxique de la protéine SOD1. Il a été démontré que l’effet mécanique d’une réduction significative de l’expression de SOD1d’environ 30% chez l’homme se produit très rapidement, quelques jours seulement après le début du traitement par Tofersen, suivi d’une baisse significative des LNF dans le liquide céphalorachidien et le sérum, avant qu’un ralentissement de la baisse du score ALSFRS-R n’apparaisse finalement aussi [16,40]. Après un suivi à plus long terme de douze mois, d’autres signaux positifs d’importance clinique ont été observés, tels que des effets sur la fonction musculaire et une stabilisation du poids.
Aux États-Unis, Tofersen a été approuvé par la FDA en avril 2023 sur la seule base de données convaincantes sur l’évolution des biomarqueurs, avec une baisse significative des valeurs NfL. En Allemagne, Tofersen a été mis à disposition dans le cadre d’un programme d’accès libre. Les premières données d’application réelles du réseau allemand de motoneurones ont confirmé de manière impressionnante les données de l’étude Tofersen, avec des données encore meilleures en termes de paramètres d’évolution clinique [41]. Dans ce contexte, il était logique que l’EMA se prononce en faveur de l’autorisation de Tofersen en février 2024.
Outre l’ASO Tofersen pour la SLA associée à la SOD1, des ASO sont actuellement développés ou font l’objet d’études, notamment contre C9orf72 et FUS [15,42]. L’ASO Jacifusen (ION363) est actuellement testé dans le cadre d’une étude multicentrique et multinationale avec deux sites d’étude (Rostock et Ulm) en Allemagne.
L’échelle révisée d’évaluation fonctionnelle de la SLA (ALSFRS-R) est un score éprouvé et bien établi pour l’évaluation des fonctions motrices des quatre régions du corps, qui représente non seulement un critère d’évaluation important pour les études cliniques, mais qui a également fait ses preuves en tant que paramètre d’évolution facile à mettre en œuvre dans la pratique clinique [43]. On s’attend à ce que l’ALSFRS-SE (SE : “self-explanatory”), qui a récemment fait l’objet d’un consensus au sein du réseau MND allemand en langue allemande avec des explications concrètes et exemplaires pour les différents items et atteintes fonctionnelles, présente un avantage supplémentaire en termes d’utilisation pratique et notamment de précision diagnostique [44].
Conclusion pour la pratique
Outre les soins standard pour toutes les formes de SLA (riluzole, prévention de l’état métabolique catabolique, y compris la mise en place d’une sonde PEG en temps voulu, ventilation non invasive précoce, voire invasive en cas d’insuffisance ventilatoire), des progrès considérables ont été réalisés, en particulier pour les formes monogéniques de SLA, grâce au développement de méthodes thérapeutiques spécifiques basées sur les gènes, notamment actuellement l’ASO. Le Tofersen est un traitement spécifique très efficace de la SLA associée à la SOD1, qui est désormais disponible en Allemagne.
Par conséquent, un diagnostic génétique de base, au moins pour les gènes les plus courants (SOD1, C9orf72, FUS, TARDP), est recommandé pour tous les patients atteints de SLA au moment du diagnostic. Nous espérons que les études à venir dans ce domaine montreront dans quelle mesure des interventions ciblées à haute teneur calorique et à effet anticatabolique peuvent influencer favorablement l’évolution de la SLA sporadique.
Messages Take-Home
- Les soins standard pour toutes les formes de SLA sont le riluzole 100 mg/d, la prévention d’un état métabolique catabolique, y compris la pose d’une sonde gastrique. La mise en place d’une sonde PEG en temps voulu, ainsi qu’une ventilation non invasive précoce, voire invasive, en cas d’insuffisance ventilatoire, sont disponibles.
- En outre, des progrès significatifs ont été réalisés, en particulier pour les formes monogéniques de SLA. Le développement de méthodes thérapeutiques spécifiques basées sur les gènes, comme actuellement les oligonucléotides antisens (ASO), en constitue la base. Le Tofersen est un traitement spécifique très efficace de la SLA associée à la SOD1.
- Un diagnostic génétique de base, au moins pour les gènes les plus courants (SOD1, C9orf72, FUS, TARDP) , est donc recommandé pour tous les patients atteints de SLA au moment du diagnostic.
Littérature :
- Duyckaerts C, Maisonobe T, Hauw JJ, Seilhean D : Charcot identifie et illustre la sclérose latérale amyotrophique. Free Neuropathol. 2. doi:10.17879/freeneuropathology-2021-3323.
- Uenal H, Rosenbohm A, Kufeldt J, et al. : Incidence et variation géographique de la sclérose latérale amyotrophique (SLA) dans le sud de l’Allemagne – Complétude du registre SLA Swabia. PLoS ONE. 2014 ; 9(4). doi:10.1371/journal.pone.0093932.
- Jun KY, Park J, Oh KW, et al : Epidémiologie de la SLA en Corée à l’aide de grandes données nationales. J Neurol Neurosurg Psychiatry. 2019;90(4) : 395-403. doi:10.1136/jnnp-2018-318974.
- Marin B, Boumédiene F, Logroscino G, et al : Variation in worldwide incidence of amyotrophic lateral sclerosis : a meta-analysis. Int J Epidemiol 2017 ; 46(1) : 57-74. doi:10.1093/ije/dyw061.
- Petri SA-O GJ Ludolph AC. “Motor neuron diseases” of the German Society of Neurology (DGN). (2524-2348 ; Electronic).
- Finsel J, Uttner I, Vázquez Medrano CR, et al. : Cognition in the course of ALS-a meta-analysis. Amyotroph Lateral Scler Front Degener. 2023 ; 24(1-2) : 2-13. doi:10.1080/21678421.2022.2101379.
- Iazzolino B, Pain D, Peotta L, et al : Validation de la classification révisée du handicap cognitif et comportemental dans la SLA. J Neurol Neurosurg Psychiatry 2019 ; 90(7) : 734-739. doi:10.1136/jnnp-2018-319696.
- Oprisan AL, Popescu BO : Dysautonomia in Amyotrophic Lateral Sclerosis. Int J Mol Sci 2023 ; 24(19). doi:10.3390/ijms241914927.
- Chiò A, Mora G, Lauria G. La douleur dans la sclérose latérale amyotrophique. Lancet Neurol 2017 ; 16(2) : 144-157. doi:10.1016/S1474-4422(16)30358-1
- Shefner JM, Al-Chalabi A, Baker MR, et al : A proposal for new diagnostic criteria for ALS. Clin Neurophysiol 2020 ; 131(8) : 1975-1978. doi:10.1016/j.clinph.2020.04.005.
- Koch JC, Petri S, Zeller D : Electrophysiologic Diagnosis of Suspected Amyotrophic Lateral Sclerosis – Consensus Recommendations of the German Network for Motor Neuron Diseases (MND-NET). Neurophysiol clinique 2024. 2024 ; 55 : 82-88.
- Zoccolella S, Mastronardi A, Scarafino A, et al : Motor-evoked potentials in amyotrophic lateral sclerosis : potential implications in detecting subclinical UMN involvement in lower motor neuron phenotype. J Neurol 2020 ; 267(12) : 3689-3695. doi:10.1007/s00415-020-10073-5.
- Shahim P, Norato G, Sinaii N, et al. Neurofilaments dans la sclérose latérale amyotrophique sporadique et familiale : A Systematic Review and Meta-Analysis. Gènes. 2024;15(4). doi:10.3390/genes15040496.
- Behzadi A, Pujol-Calderón F, Tjust AE, et al : Les neurofilaments peuvent différencier les sous-groupes de la SLA et la SLA des mimiques de diagnostic communes. Sci Rep 2021 ; 11. doi:10.1038/s41598-021-01499-6.
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, et al : Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med 2022 ; 28(1) : 104-116. doi:10.1038/s41591-021-01615-z.
- Miller TM, Cudkowicz ME, Genge A, et al : Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. Publié en ligne le 22 septembre 2022. doi:10.1056/NEJMoa2204705.
- Ruf WP, Boros M, Freischmidt A, et al : Spectrum and frequency of genetic variants in sporadic amyotrophic lateral sclerosis. Brain Commun 2023 ; 5(3). doi:10.1093/braincomms/fcad152.
- Braak H, Brettschneider J, Ludolph AC, et al : Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 2013 ; 9(12) : 708-714. doi:10.1038/nrneurol.2013.221.
- Brettschneider J, Del Tredici K, Toledo JB, et al : Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013 ; 74(1) : 20-38. doi:10.1002/ana.23937.
- Neumann M : Neuropathologie moléculaire des protéinopathies TDP-43. Int J Mol Sci 2009 ; 10(1) : 232-246. doi:10.3390/ijms10010232.
- Saberi S, Stauffer JE, Schulte DJ, Ravits J : “Neuropathologie de la sclérose latérale amyotrophique et de ses variantes”. Neurol Clin 2015 ; 33(4) : 855-876. doi:10.1016/j.ncl.2015.07.012.
- Braak H, Braak E : Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 1995 ; 16(3) : 271-278-284.
- Braak H, Ludolph AC : Neumann M, Ravits J, Del Tredici K. Les changements pathologiques du TDP-43 dans les cellules de Betz diffèrent de ceux observés dans les motoneurones α bulbaires et spinaux dans la sclérose latérale amyotrophique sporadique. Acta Neuropathol (Berl) 2017 ; 133(1) : 79-90. doi:10.1007/s00401-016-1633-2.
- Eisen A, Braak H, Del Tredici K, et al : Cortical influences drive amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2017 ; 88(11) : 917-924. doi:10.1136/jnnp-2017-315573.
- Ludolph AC, Emilian S, Dreyhaupt J, et al : Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J Neurol Neurosurg Psychiatry 2020 ; 91(9) : 991-998. doi:10.1136/jnnp-2020-323331.
- Menon P, Kiernan MC, Vucic S : L’hyperexcitabilité corticale précède le dysfonctionnement des neurones moteurs inférieurs dans la SLA. Clin Neurophysiol 2015 ; 126(4) : 803-809. doi:10.1016/j.clinph.2014.04.023.
- Prasad A, Bharathi V, Sivalingam V, et al : Mécanismes moléculaires du déphasage et de la pathologie du TDP-43 dans la sclérose latérale amyotrophique. Front Mol Neurosci 2019;12. doi:10.3389/fnmol.2019.00025.
- Gosset P, Camu W, Raoul C, Mezghrani A : Prionoïdes dans la sclérose latérale amyotrophique. Brain Commun 2022;4(3). doi:10.1093/braincomms/fcac145.
- Hardiman O, Al-Chalabi A, Chio A, et al : Sclérose latérale amyotrophique. Nat Rev Dis Primer 2017 ; 3:17071. doi:10.1038/nrdp.2017.71.
- Masrori P, Van Damme P : Amyotrophic lateral sclerosis : a clinical review. Eur J Neurol 2020 ; 27(10) : 1918-1929. doi:10.1111/ene.14393.
- Eisen A, Vucic S, Mitsumoto H : Histoire de la SLA et des théories concurrentes sur la pathogenèse : chapitre du manuel de l’IFCN. Clin Neurophysiol Pract 2023 ; 9 : 1-12. doi:10.1016/j.cnp.2023.11.004.
- Rosen DR, Siddique T, Patterson D, et al : Des mutations dans le gène de la superoxyde dismutase Cu/Zn sont associées à la sclérose latérale amyotrophique familiale. Nature 1993 ; 362(6415) : 59-62. doi:10.1038/362059a0.
- Bensimon G, Lacomblez L, Meininger V, Group the AS : A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. http://dx.doi.org/10.1056/NEJM199403033300901.
- Hinchcliffe M, Smith A : Riluzole : real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis 2017;7 : 61-70. doi:10.2147/DNND.S135748.
- Miller RG, Mitchell JD, Moore DH : Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND).
Cochrane Database Syst Rev 2012 ; 2012(3).
doi:10.1002/14651858.CD001447.pub3. - Dupuis L, Pradat PF, Ludolph AC, Loeffler JP : Métabolisme énergétique dans la sclérose latérale amyotrophique. Lancet Neurol 2011 ; 10(1) : 75-82. doi:10.1016/S1474-4422(10)70224-6.
- Cui F, Sun L, Xiong J, et al : Effets thérapeutiques de la gastrostomie endoscopique percutanée sur la survie des patients atteints de sclérose latérale amyotrophique : une méta-analyse. PLoS ONE 2018 ; 13(2). doi:10.1371/journal.pone.0192243.
- Dorst J, Ludolph AC : Ventilation non invasive dans la sclérose latérale amyotrophique. Ther Adv Neurol Disord 2019 ; 12 : 1756286419857040. doi:10.1177/1756286419857040.
- Radunovic A, Annane D, Rafiq MK, et al : Ventilation mécanique pour la sclérose latérale amyotrophique/maladie des neurones moteurs. Cochrane Database Syst Rev 2017 ; 2017(10).
doi:10.1002/14651858.CD004427.pub4. - Miller T, Cudkowicz M, Shaw PJ, et al : Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2020 ; 383(2) : 109-119. doi:10.1056/NEJMoa2003715.
- Wiesenfarth M, Dorst J, Brenner D, et al. : Effects of tofersen treatment in patients with SOD1-ALS in a “real-world” setting – a 12-month multicenter cohort study from the German early access program. eClinicalMedicine. 2024 ; 69. doi:10.1016/j.eclinm.2024.102495.
- Meijboom KE, Brown RH : Approches de la thérapie de modulation génétique pour la SLA. Neurotherapeutics 2022;19(4) : 1159-1179. doi:10.1007/s13311-022-01285-w.
- Cedarbaum JM, Stambler N, Malta E, et al. : The ALSFRS-R : a revised ALS functional rating scale that incorporates assessments of respiratory function. Groupe d’étude BDNF ALS (phase III). J Neurol Sci 1999 ; 169(1-2) : 13-21. doi:10.1016/s0022-510x(99)00210-5.
- Maier A, Boentert M, Reilich P, et al : ALSFRS-R-SE : an adapted, annotated, and self-explanatory version of the revised amyotrophic lateral sclerosis functional rating scale. Neurol Res Pract 2022 ; 4. doi:10.1186/s42466-022-00224-6.
| Première publication dans neuro aktuell 2024 ; 38(8) : 30-35. |
HAUSARZT PRAXIS 2024; 19(11): 12–17









