Le traitement prophylactique par des concentrés de facteurs est devenu la norme dans le traitement des patients atteints d’hémophilie A sévère. Les concentrés conventionnels de facteur VIII et de facteur IX ont une demi-vie relativement courte et doivent donc souvent être administrés par voie intraveineuse. Depuis quelques mois, les produits de facteur VIII et de facteur IX à demi-vie prolongée sont tous deux autorisés en Suisse. Diverses options thérapeutiques alternatives pour le traitement de l’hémophilie pourraient être mises sur le marché dans les années à venir.
Bien que le terme “hémophilie” recouvre en principe tous les troubles congénitaux de la coagulation qui entraînent une tendance accrue aux saignements, il décrit au sens strict deux troubles définis de la coagulation sanguine : d’une part, le déficit en facteur VIII de la coagulation (hémophilie A) et, d’autre part, le déficit en facteur IX de la coagulation sanguine (hémophilie B). Ces deux troubles sont transmis sur le mode récessif lié au chromosome X. Outre les hémophilies héréditaires, il existe également des formes acquises (hémophilies auto-immunes), très rares, mais qui ne seront pas abordées ici.
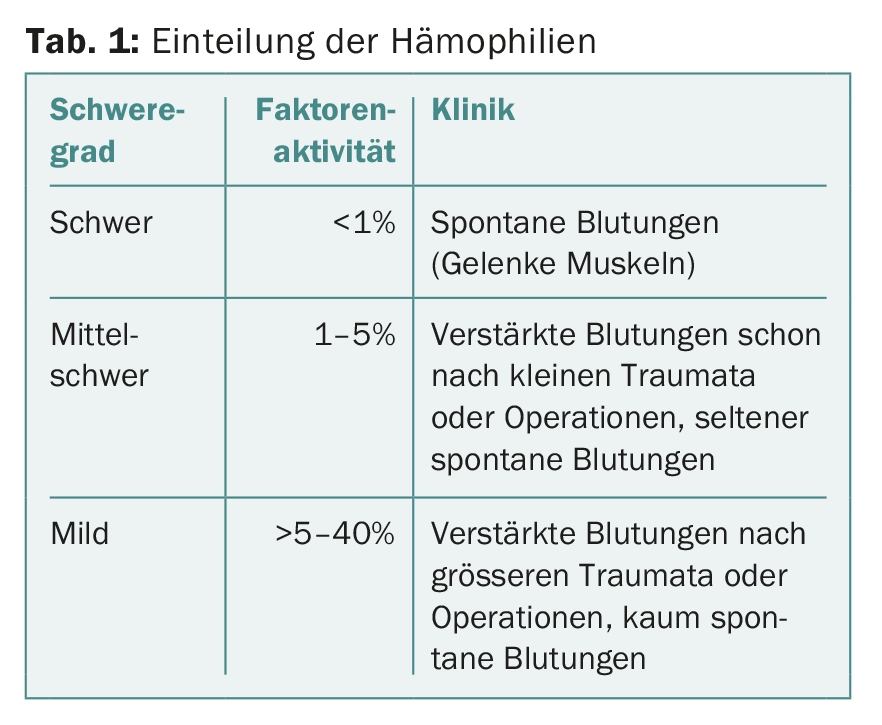
Dans les hémophilies héréditaires, l’activité résiduelle du facteur VIII ou IX est relativement bien corrélée à la présentation clinique. Cela permet de classer les hémophilies A et B en fonction de l’activité résiduelle des facteurs VIII et IX respectivement (tableau 1).
Traitement de l’hémophilie
L’invention, il y a plus de 50 ans, du traitement substitutif intraveineux à base de concentrés de facteurs issus du plasma humain a permis de réduire considérablement la mortalité et la morbidité de l’hémophilie en diminuant les épisodes hémorragiques. Cependant, cette réussite a été fortement ternie par la transmission de maladies virales telles que le VIH et l’hépatite C dans les années 1970 et 1980. La mise en œuvre de meilleures méthodes de dépistage et d’inactivation virale ainsi que le développement de préparations de facteurs recombinants ont toutefois permis d’améliorer considérablement la sécurité des préparations, de sorte qu’aucune transmission de virus n’a été signalée depuis 1990. Aujourd’hui, le principal risque du traitement par les concentrés de facteurs est l’apparition d’inhibiteurs, qui sont observés chez environ 20 à 30% des patients atteints d’hémophilie A sévère (un peu moins souvent pour l’hémophilie B). Comme leur nom l’indique, ces inhibiteurs inhibent les facteurs de coagulation apportés et rendent le traitement de substitution inefficace. Il est toutefois possible, dans de nombreux cas, de se débarrasser des inhibiteurs grâce à des thérapies complexes de tolérance immunitaire.
Chez les patients atteints d’hémophilie sévère, la norme est ce que l’on appelle la “prophylaxie”. Dans ce cas, des concentrés de facteurs sont (en général) administrés par voie intraveineuse deux à trois fois par semaine de manière à éviter en grande partie les saignements (articulaires) spontanés en augmentant les concentrations plasmatiques du facteur de coagulation concerné. La fréquence d’injection dépend en fin de compte de la demi-vie du facteur de coagulation concerné (facteur VIII env. 12 h, facteur IX env. 18 h) [1].
Les injections intraveineuses relativement fréquentes sont vécues comme pénibles par de nombreux patients et, en fin de compte, la normalisation continue du système de coagulation n’est pas atteinte.
Nouvelles préparations de facteurs
Il existe néanmoins un certain potentiel d’amélioration dans le développement de nouvelles options thérapeutiques pour l’hémophilie : allongement de la demi-vie (donc moins d’injections et meilleure protection), formes d’administration alternatives (par voie orale ou sous-cutanée plutôt qu’intraveineuse), immunogénicité moindre (moins de formation d’inhibiteurs) et, en principe, meilleure efficacité.
Les techniques de prolongation de la demi-vie sont certainement les plus avancées. En principe, les facteurs de coagulation recombinants sont modifiés de manière à ce qu’ils soient dégradés moins rapidement. Diverses préparations de ce type à “longue durée d’action” ont obtenu une autorisation de mise sur le marché en Suisse au cours des derniers mois. Les approches utilisées sont d’une part la modification chimique des facteurs (couplage à des polyéthylèneglycols (PEGylation)), la fusion avec d’autres protéines (région Fc de l’immunoglobuline G, albumine recombinante) ou la modification ciblée de la séquence protéique. Pour être complet, il convient de mentionner que certaines préparations de facteur VIII classique recombinant sont également apparues sur le marché ces dernières années et que, grâce à des procédés de fabrication optimisés (p. ex. glycolisation optimisée), elles ont tendance à avoir des demi-vies plus longues que leurs prédécesseurs. Ces préparations ne sont toutefois pas considérées comme des “demi-vies prolongées” au sens strict.
Les méthodes utilisées pour obtenir une demi-vie prolongée sont brièvement présentées ci-dessous.
PEGylation : cette technique consiste à attacher une à plusieurs molécules de PEG de 20 à 60 kDa à une molécule de facteur VIII ou IX. Les différents fabricants utilisent à cet égard des concepts différents. Il est essentiel que les molécules de PEG soient attachées à la molécule de facteur de telle sorte que son action hémostasique ne soit pas compromise.
L’enrobage par des molécules de PEG rend les molécules de facteur moins accessibles aux protéases, et la PEGylation entraîne une augmentation de la taille et une meilleure solubilité dans l’eau. Tout cela conduit à une demi-vie prolongée des facteurs PEGylés [2].
Technologie des protéines de fusion : la fusion d’une protéine de coagulation avec une autre protéine, qui a naturellement une demi-vie beaucoup plus longue dans la circulation sanguine, permet finalement d’augmenter la demi-vie de la protéine de coagulation elle-même.
Concrètement, la région Fc de l’immunoglobuline G (IgG-Fc) ou l’albumine recombinante sont utilisées pour la fusion. Les IgG-Fc et l’albumine ont toutes deux des demi-vies très longues, car elles sont protégées de la dégradation lysosomale par une sorte de mécanisme de recyclage [3].
Modification de séquence spécifique au site : cette technique consiste à modifier la molécule de facteur VIII de manière à ce qu’elle présente une affinité de liaison nettement plus élevée avec sa molécule porteuse, le facteur von Willebrand. Le facteur von Willebrand a une demi-vie légèrement plus longue que celle du facteur VIII, soit environ 15 heures. En raison de la forte liaison de la molécule de facteur VIII à la molécule de facteur von Willebrand , la demi-vie du facteur VIII se rapproche de celle du facteur von Willebrand [4].
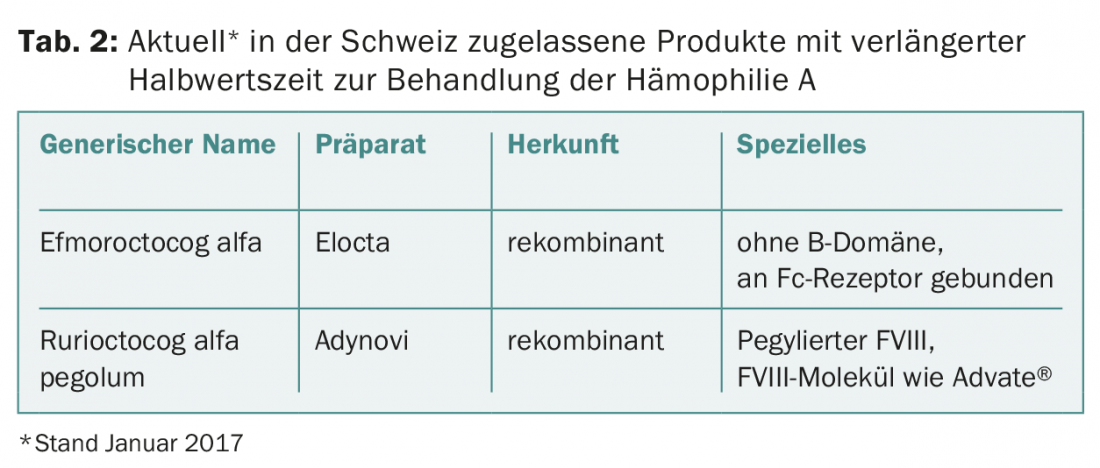
Prolongation de la demi-vie des produits FVIII : La demi-vie du facteur VIII est finalement limitée par le facteur von Willebrand [5]. En effet, le facteur von Willebrand, en tant que protéine porteuse, protège le facteur VIII contre la dégradation protéolytique. Toutes les mesures décrites ci-dessus permettent donc au maximum de prolonger d’environ 1,5 fois la demi-vie du facteur VIII (environ 12 h). Ce n’est pas beaucoup, mais cela représente déjà un progrès mesurable sur le plan clinique. Par exemple, si un patient a besoin de trois injections par semaine avec un produit conventionnel, il pourrait atteindre les mêmes niveaux de vallée avec deux injections par semaine avec un produit à demi-vie prolongée. En revanche, si le même patient maintenait sa fréquence d’injection de trois fois par semaine avec le nouveau produit, il pourrait obtenir des niveaux de vallée nettement plus élevés. Cela serait par exemple utile si, malgré une prophylaxie régulière, il avait jusqu’à présent des hémorragies de rupture [6]. Les produits de facteur VIII à demi-vie prolongée actuellement autorisés en Suisse sont présentés dans le tableau 2 .
Allongement de la demi-vie des produits à base de facteur IX : Le facteur IX normal de coagulation du sang a une demi-vie d’environ 18 à 20 heures. Contrairement au facteur VIII, le facteur IX n’est pas lié à une protéine porteuse. Les mesures décrites ci-dessus ont donc un effet beaucoup plus impressionnant sur le facteur IX que sur le facteur VIII : la PEGylation et les protéines de fusion ont toutes deux pour effet de multiplier la demi-vie par 5, c’est-à-dire de la porter à environ 90 heures.
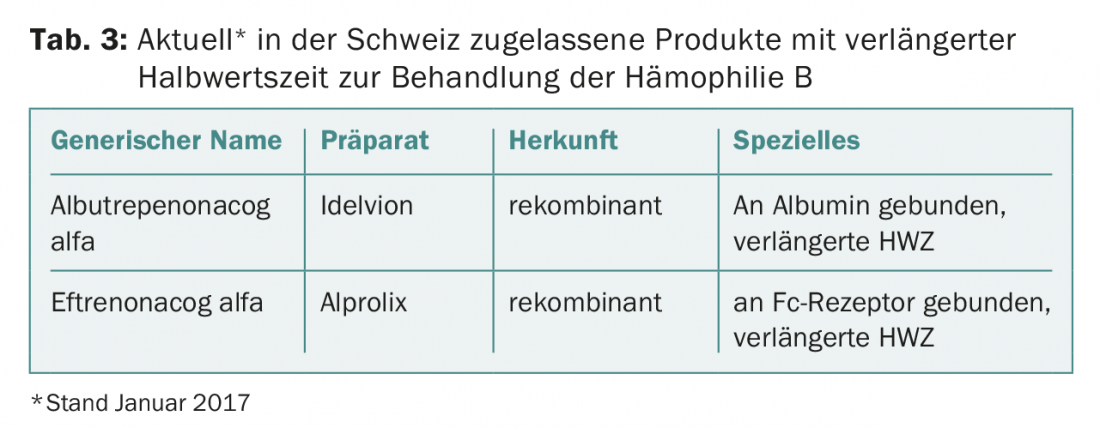
Ce résultat impressionnant ouvre la voie à de nouvelles stratégies thérapeutiques. Les préparations de facteur IX à demi-vie prolongée ne doivent plus être appliquées que toutes les une à deux semaines pour la prophylaxie (les préparations de facteur IX normales environ deux fois par semaine) et il est malgré tout possible d’obtenir des taux de vallée encore plus élevés [7]. Les produits de facteur IX à demi-vie prolongée actuellement autorisés en Suisse sont présentés dans le tableau 3.
Approches thérapeutiques alternatives
Outre les méthodes susmentionnées, qui ont toutes pour objectif d’optimiser le traitement de substitution par des concentrés de facteurs, de nombreuses approches thérapeutiques alternatives ont été mises en place. Il s’agit d’essayer d’obtenir, par des manipulations ciblées du système de coagulation, une activation suffisante de la coagulation malgré l’hémophilie existante, afin d’éviter au moins les saignements spontanés. Les avantages potentiels d’un écart par rapport au concept classique de substitution de facteurs sont, entre autres, d’éviter le problème du développement d’inhibiteurs et de permettre des formes d’administration alternatives.
L’une de ces approches alternatives consiste à bloquer l’inhibition physiologique de la coagulation. Concrètement, les recherches visent à inhiber ou à réguler à la baisse le TFPI (Tissue Factor Pathway Inhibitor) ou l’antithrombine. Le fitusiran, par exemple, est une molécule qui peut cibler l’antithrombine. Cette molécule d’interférence ARN, qui peut être appliquée par voie sous-cutanée, inhibe spécifiquement la formation d’antithrombine. Cela se traduit dans le système de coagulation par une meilleure génération de thrombine, ce qui permet un meilleur contrôle des saignements chez les patients hémophiles [8]. Le fitusiran a déjà été testé cliniquement avec succès (phase 1/2).
Une autre substance intéressante, qui suit jusqu’à présent avec succès un programme d’études cliniques, est l’anticorps bi-spécifique emicizumab. Cet anticorps imite dans une certaine mesure la fonction du facteur VIII activé en se liant simultanément aux facteurs de coagulation IX et X, ce qui entraîne une activation accrue du facteur X et donc une coagulation accélérée du sang. L’emicizumab peut être injecté par voie sous-cutanée et il est également efficace en présence d’inhibiteurs du facteur VIII. Cependant, l’anticorps n’atteint pas la même efficacité biologique que le facteur de coagulation naturel VIII [9].
Une autre approche thérapeutique toujours en cours est la thérapie génique. Des études ont permis d’améliorer l’activité endogène du facteur IX à long terme chez des patients atteints d’hémophilie B (sévère), mais aucune normalisation de l’activité du facteur IX n’a été obtenue à ce jour. Dans le cas de l’hémophilie A, la thérapie génique n’est pas encore techniquement possible [9].
Conclusion
Les nombreux nouveaux produits qui ont été lancés ces derniers mois et qui le seront dans les années à venir sont très prometteurs. Certaines des préparations mentionnées ont tout à fait le potentiel de révolutionner le traitement de l’hémophilie. Il n’est toutefois pas encore clair si les coûts potentiellement élevés, notamment des approches thérapeutiques alternatives discutées ci-dessus, pourraient influencer l’utilisation clinique.
En raison des nombreuses options dont nous disposons dans les pays développés pour traiter l’hémophilie, nous ne devons pas oublier qu’un grand nombre de ces patients n’ont toujours pas un accès suffisant aux concentrés de facteurs.
Littérature :
- Ljung R : Aspects du traitement prophylactique de l’hémophilie. Journal de la thrombose. 2016;14(Suppl 1) : 30.
- Ivens IA, et al : PEGylated therapeutic proteins for haemophilia treatment : a review for haemophilia caregivers. Haemophilia : the official journal of the World Federation of Hemophilia. 2013;19(1) : 11-20.
- Powell JS : Concentrats de facteurs de clotting à action prolongée pour l’hémophilie. Journal of thrombosis and haemostasis : JTH. 2015;13 Suppl 1 : S167-75.
- Buyue Y, Liu T, et al : A single chain variant of factor VIII Fc fusion protein retains normal in vivo efficacy but exhibits altered in vitro activity. PloS one. 2014;9(11) : e113600.
- Pipe SW, et al. : Life in the shadow of a dominant partner : the FVIII-VWF association and its clinical implications for hemophilia A. Blood. 2016.
- Tiede A : Half-life extended factor VIII for the treatment of hemophilia A. Journal of thrombosis and haemostasis : JTH. 2015;13 Suppl 1 : S176-9.
- Nazeef M, Sheehan JP : New developments in the management of moderate-to-severe hemophilia B. Journal of blood medicine. 2016;7 : 27-38.
- Sehgal A, et al : Un ARNi thérapeutique ciblant l’antithrombine pour rééquilibrer le système de coagulation et promouvoir l’hémostase dans l’hémophilie. Nature medicine. 2015;21(5) : 492-7.
- Shima M, Hanabusa H, Taki M, et al. : Fonction mimétique du facteur VIII de l’anticorps bispécifique humain dans l’hémophilie A. The New England journal of medicine. 2016;374(21) : 2044-53.
InFo ONKOLOGIE & HÄMATOLOGIE 2017 ; 5(1) : 5-9









