Les sarcomes sont des tumeurs malignes rares du tissu conjonctif. Le traitement est multidisciplinaire et doit être effectué dans des centres de sarcome. En Suisse, toutes les disciplines du sarcome se sont organisées à l’échelle nationale (www.sarcoma.ch). C’est ici que sont publiés les guides thérapeutiques, que sont mis en place un registre, une étude de cohorte et une banque de tissus. L’ablation chirurgicale complète de la tumeur constitue la mesure thérapeutique la plus importante, tant pour les tumeurs osseuses que pour les tumeurs des tissus mous. Dans l’ensemble, on opère autant que possible en préservant les extrémités, mais en cas de tumeur osseuse, la reconstruction se fait de préférence avec une endoprothèse (tumorale modulaire) ou en combinaison avec une allogreffe. La radiothérapie est souvent utilisée pour les tumeurs des tissus mous, en particulier lorsque la tumeur est adjacente à des structures neurovasculaires, afin d’améliorer le contrôle local. Une chimiothérapie systémique additive, généralement à base de doxorubicine, est principalement utilisée pour améliorer le pronostic des ostéosarcomes, des sarcomes d’Ewing, des rhabdomyosarcomes et des métastases.
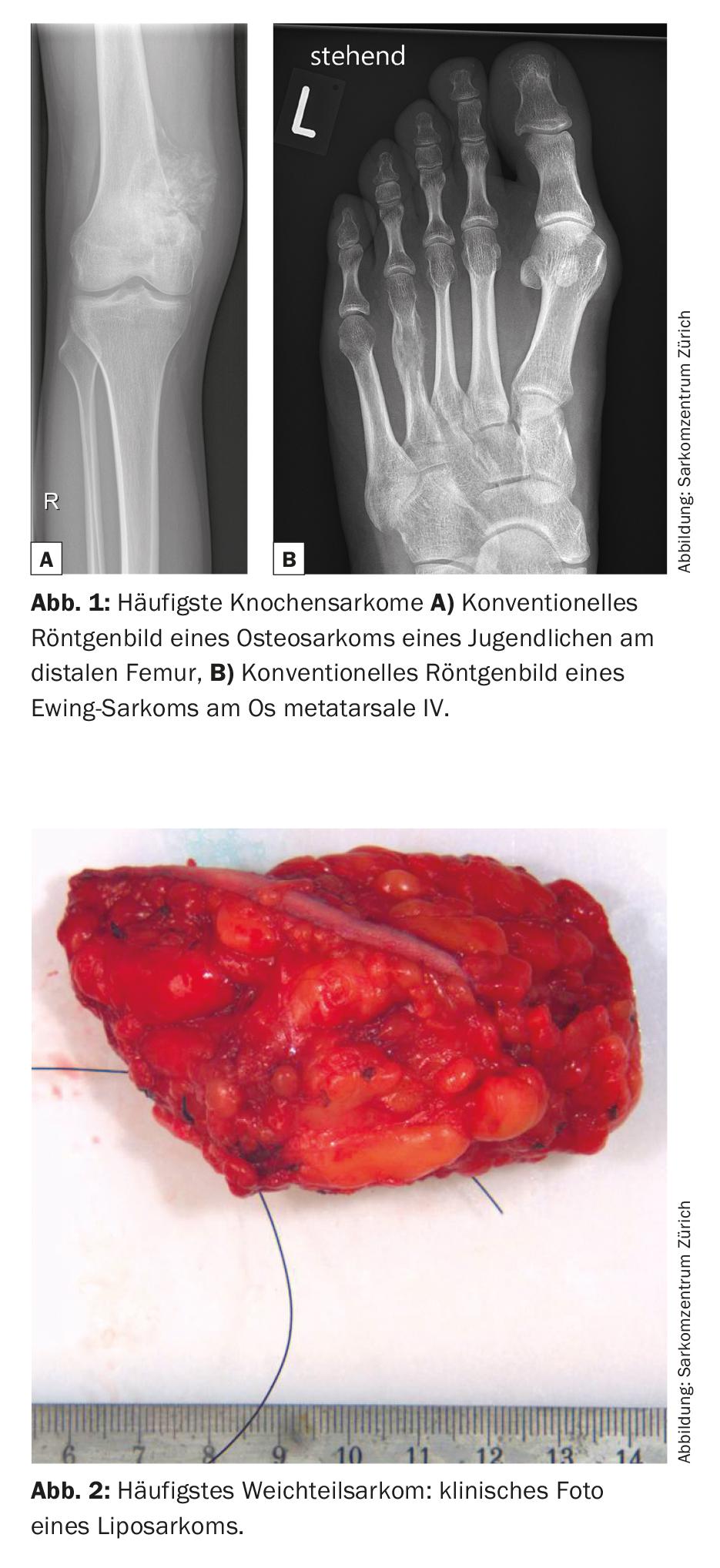
Les sarcomes sont des tumeurs malignes rares du tissu conjonctif qui représentent 6% de tous les cancers de l’enfant [1]. Il existe de nombreux sous-types, plus de 50 au total, qui peuvent généralement être divisés en sarcomes des os et des tissus mous. L’ostéosarcome métaphysaire et le sarcome d’Ewing diaphysaire sont les sarcomes osseux les plus fréquents, tandis que le liposarcome est l’un des sarcomes des tissus mous les plus courants (figures 1 et 2).
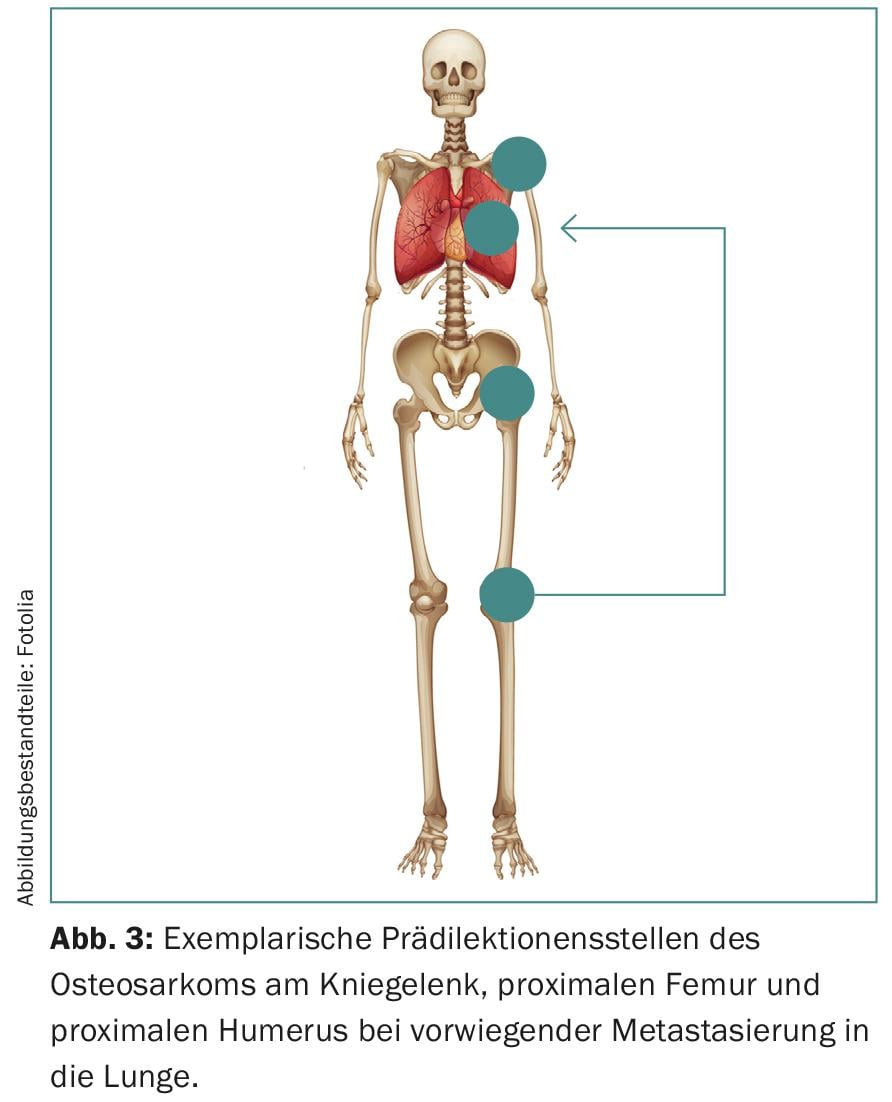
En outre, les sarcomes présentent des prédictions pour certains groupes d’âge et structures anatomiques (figure 3). Ils se manifestent principalement au niveau des extrémités et du squelette axial, avec une répartition bimodale en fonction de l’âge. La région des genoux est particulièrement touchée chez les adolescents, bien que la tête, le cou et l’abdomen puissent également être atteints.
Diagnostic
Dans la plupart des cas, le diagnostic se fait sur la base d’une découverte fortuite, même si un gonflement et des douleurs peuvent parfois être observés. Ces dernières ont classiquement un caractère local, progressif, à prédominance nocturne et non modifiable par les AINS (anti-inflammatoires non stéroïdiens) [2]. L’examen clinique doit d’abord consister à inspecter et à palper localement le tissu tumoral avec la peau qui le recouvre et les ganglions lymphatiques voisins. Ensuite, une recherche de métastases peut être étendue à la thyroïde, à l’abdomen, à la prostate ou aux seins. Une symptomatologie B systémique est très rare.
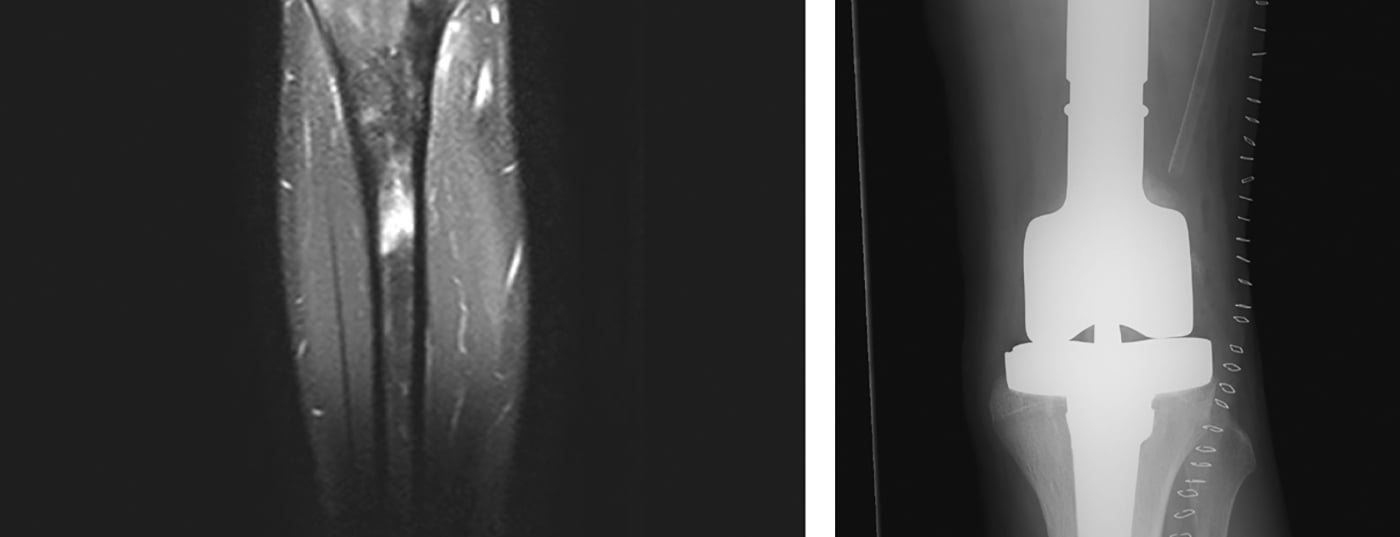
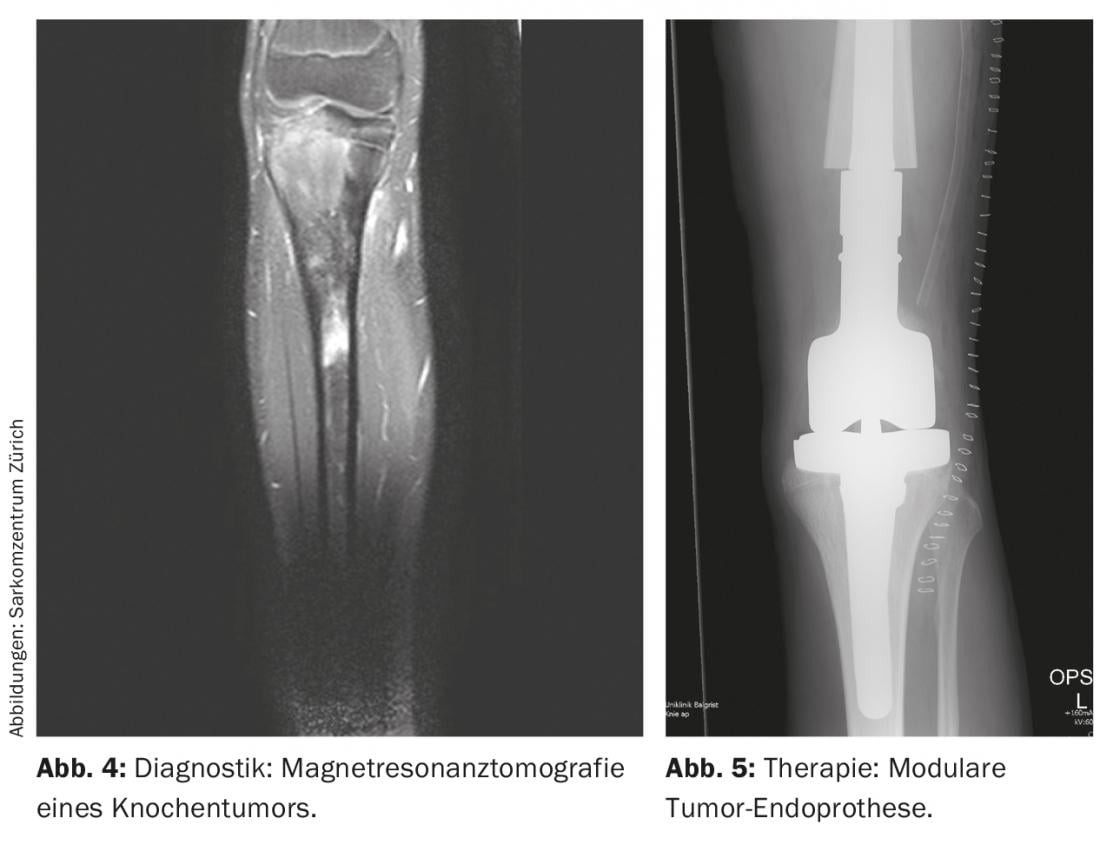
La localisation de la tumeur est effectuée à l’aide de l’imagerie radiologique, qui comprend des radiographies locales et thoraciques en deux plans et, le cas échéant, un scanner (tomodensitométrie), une IRM (imagerie par résonance magnétique) locale (figure 4) et une scintigraphie du squelette au Tc (technétium) systémique ou, à défaut, un PET-CT. En règle générale, il convient de noter que les radiographies conventionnelles des os longs doivent toujours montrer les deux articulations adjacentes. En outre, les poumons sont également examinés car ils sont le plus souvent touchés par les métastases, dans environ 10% des cas. A l’examen, les ostéosarcomes et les sarcomes d’Ewing présentent classiquement un éperon périosté appelé triangle de Codman, et des calcifications périostées en pelure d’oignon. En principe, la taille (≤ ou >10 cm), la localisation (épi- ou sous-fasciale) et le statut métastatique de la tumeur doivent être consignés. En outre, les analyses chimiques de routine en laboratoire et le dosage de la lactate déshydrogénase et de la phosphatase alcaline sont utiles pour exclure les maladies généralisées et évaluer la charge de morbidité, même si elles sont relativement peu spécifiques.
Enfin, une biopsie guidée par scanner ou US, souvent peu invasive de nos jours, peut fournir des informations sur le sous-type spécifique de sarcome grâce à des analyses microscopiques, immunologiques et génétiques moléculaires. On veille à ce que le trajet de la biopsie soit choisi de manière à ce qu’il puisse être retiré in toto lors d’une éventuelle opération ultérieure. Si une biopsie à l’emporte-pièce donne des résultats équivalents, une biopsie ouverte est effectuée. Il s’agit d’éviter les hématomes dus au risque de dissémination en procédant à une fermeture du fascia musculaire ou à une insertion de redon. En intra-intervention, un prélèvement frais sur glace est toujours réalisé. En outre, par mesure de sécurité, un examen bactériologique doit être effectué. C’est pourquoi il est déconseillé d’administrer des antibiotiques avant l’intervention, afin de ne pas compliquer les résultats des examens et le traitement en fonction de la résistance.
Sur la base du bloc de tissu paraffiné, on procède à la classification histologique, pour laquelle il existe plusieurs possibilités. Selon la classification, des examens spécifiques de biologie moléculaire sont ensuite effectués. La classification en six stades (IA-IIIB) selon Enneking (Musculoskeletal Tumor Society) tient compte des métastases (non vs oui), du grade histologique (faible vs élevé) et de la taille anatomique de la tumeur (intra- vs extra-compartimentale). La classification en sept stades (IA-IVB) de l’AJCC (American Joint Commission on Cancer) est un peu plus détaillée et indique l’atteinte des ganglions lymphatiques.
Thérapie
Les thérapies élaborées dans les centres de sarcome hautement spécialisés donnent des résultats relativement bons dans le traitement à long terme des sarcomes localisés, avec des taux de survie à 5 ans pouvant atteindre 75% [3]. En revanche, les taux de survie pour les sarcomes métastatiques ne sont que d’environ 25%. Ces résultats ayant atteint un plateau depuis plus d’une décennie, les sarcomes continuent de faire l’objet de divers projets de recherche internationaux afin d’acquérir une meilleure compréhension de la génétique moléculaire et de définir ainsi de nouvelles options thérapeutiques.
Le traitement nécessite une équipe interdisciplinaire composée de chirurgiens orthopédiques spécialisés dans les tumeurs, de radiologues, d’oncologues, de radiologues et de pathologistes. En fonction de ses entités démographiques, chaque patient, mais aussi chaque sous-type de sarcome, devrait, à l’instar des néoplasmes hématopoïétiques, faire l’objet d’un traitement unique, multidisciplinaire, adapté aux changements biologiques et souvent multimodal. Dans l’ensemble, l’objectif thérapeutique curatif en cas de tumeur localisée est l’excision chirurgicale complète de la tumeur. Dans le cas des sarcomes osseux en particulier, une chimiothérapie et une radiothérapie sont utilisées de manière additive pour réduire les (micro-)métastases et les récidives locales. Les sarcomes des tissus mous sont très souvent traités par une thérapie combinée, la radiothérapie étant effectuée soit avant, soit après la chirurgie. Une approche palliative doit être envisagée dès lors que des métastases sont présentes au moment du diagnostic, ce qui concerne environ 10% des patients. Des lignes directrices pour l’évaluation, le diagnostic, les options thérapeutiques et le suivi ont été établies par le Swiss National Sarcoma Advisory Board et publiées sur le site Internet www.sarcoma.ch.
Opération
Depuis son introduction il y a plus de 30 ans, l’ablation chirurgicale complète de la tumeur s’est imposée comme la principale option thérapeutique (Fig. 2), et présente la meilleure “therapy response” de toutes les modalités thérapeutiques. Elle peut être utilisée comme traitement unique, en particulier pour les tumeurs des tissus mous de petite taille (<5 cm), complètement retirées et de faible grade [4]. La nature de la marge de résection est déterminante pour le taux de récidive, qui est de 100% pour une marge intralésionnelle, <50% pour une marge marginale et <10% pour une marge large (>1 cm). Si l’ablation complète des tissus tumoraux n’est pas réalisée lors de l’opération primaire, il convient d’envisager une opération secondaire dans le sens d’une résection ultérieure, plutôt que d’envisager d’autres options thérapeutiques de substitution. En outre, la métastasectomie curative reste controversée.
Le choix de la méthode chirurgicale la plus appropriée repose avant tout sur la classification et la localisation de la tumeur. En règle générale, les techniques chirurgicales de conservation des membres sont privilégiées chaque fois que cela est possible. Celles-ci incluent une résection initiale de la tumeur en bloc suivie d’une reconstruction. L’objectif est de réaliser une résection tumorale équivalente à une amputation classique tout en conservant une fonction aussi physiologique que possible. En ce qui concerne les localisations tumorales axiales, la navigation guidée par scanner peut être utile pour épargner les structures neurovasculaires importantes, en particulier dans les opérations pelviennes difficiles. Néanmoins, le pronostic des tumeurs axiales est moins bon que celui des extrémités en raison de la difficulté à les réséquer [5].
On utilise généralement des allogreffes et/ou des endoprothèses modulaires (tumorales) (Fig. 5) [6]. Les allogreffes sont devenues plus disponibles ces dernières années grâce aux banques d’os, fournissent un revêtement adéquat des tissus mous et conduisent à une ostéointégration dans le corps après plusieurs années. Cependant, il n’y a pas seulement un risque accru de fracture, mais aussi un risque de pseudarthrose en raison de l’ossification de la zone hôte de la greffe, qui peut durer jusqu’à un an. Les endoprothèses sont principalement utilisées pour les tumeurs métaphysaires nécessitant une résection articulaire et/ou épiphysaire, afin de préserver l’amplitude des mouvements et de compenser les différences de longueur des jambes. Elles offrent l’avantage d’une mise en charge complète postopératoire relativement rapide avec une bonne amplitude de mouvement. Cependant, il existe les inconvénients habituels des prothèses, qui peuvent se desserrer chez environ 5 à 10 % des patients. A cet égard, ils montrent des taux de survie à 5 et 10 ans d’environ 80% et 70% respectivement. Par rapport à l’implantation d’une prothèse seule, un composite allogreffe-prothèse présente l’avantage d’un meilleur ancrage dans les tissus mous avec un risque de descellement moindre, et permet par ailleurs de combiner les autres avantages et inconvénients des deux méthodes.
L’amputation est pratiquée dans moins de 10% des cas et est aujourd’hui utilisée en dernier recours, car elle ne permet pas d’obtenir de meilleurs taux de survie qu’une chirurgie de conservation des membres réalisée de manière adéquate [7]. Elle constitue une option thérapeutique valable, en particulier pour les récidives non reconstructibles, affectant les structures neurovasculaires et limitant ainsi la fonction. A cet égard, les prothèses actuelles offrent de bonnes et nombreuses possibilités, en particulier pour les moignons d’amputation les plus distaux possibles. En outre, une plastie d’inversion de Van Ness est une amputation intercalaire raccourcissant le membre, dans laquelle la jambe, y compris le pied, est fixée au membre proximal. Cela permet à l’articulation supérieure de la cheville d’assumer la fonction du genou et de mieux contrôler une prothèse.
Chimiothérapie
Une chimiothérapie systémique peut être administrée en complément de la chirurgie, soit en préopératoire, soit en postopératoire, ou les deux selon le type de tumeur. Celle-ci consiste le plus souvent en une combinaison à base de doxorubicine [8], la doxorubicine interférant avec la synthèse de l’ADN et entraînant l’apoptose par formation de radicaux libres. Dans le cas d’un ostéosarcome, elle consiste généralement en plusieurs cycles de doxorubicine, de méthotrexate et de cisplatine. Outre les ostéosarcomes, les sarcomes d’Ewing, les rhabdomyosarcomes et les métastases sont souvent traités par chimiothérapie. Cependant, leur effet thérapeutique et donc pronostique est controversé, en particulier pour les sarcomes des tissus mous. En raison des effets secondaires non négligeables tels que la néphro- ou la cardiotoxicité, une analyse du rapport bénéfice/risque doit donc être effectuée au cas par cas. En outre, il est préférable de l’utiliser dans le cadre d’études cliniques.
L’administration peut se faire en préopératoire, en néoadjuvant, ou en postopératoire, en adjuvant, pendant environ sept mois. L’avantage de l’utilisation néoadjuvante réside dans la réduction de la masse tumorale, le traitement des micrométastases invisibles radiologiquement, et dans la pertinence du pronostic. Les micrométastases sont particulièrement présentes dans les ostéosarcomes, jusqu’à 80% des cas. Le pronostic dépend de manière décisive de la réponse à la chimiothérapie et se reflète dans le taux de nécrose histologique (≥90% = bon vs. <90% = mauvais) selon Salzer-Kuntschik.
Radiothérapie
La radiothérapie constitue une autre option thérapeutique, en particulier pour les sarcomes des tissus mous. Elle permet de réduire le taux de récidive, qui est inférieur à 10%. On ne sait toujours pas si cela a également une influence sur la formation de métastases et le pronostic. Elle est principalement utilisée pour les sarcomes de grande taille, profonds, de haut grade, incomplètement excisés, englobant les structures neurovasculaires, affectant les tissus mous et métastasés [9]. D’une manière générale, elle provoque, par le biais des radicaux libres, des cassures double brin toxiques de l’ADN cellulaire (acide désoxyribonucléique) et donc l’apoptose. L’utilisation préopératoire est souvent privilégiée. Cela permet d’une part d’obtenir une dose thérapeutique plus faible, d’environ 50 Gy (Gray), en raison du contourage plus détaillé et du volume plus faible, et d’autre part d’obtenir un meilleur résultat fonctionnel à long terme. Cependant, il existe également un risque accru de troubles de la cicatrisation postopératoire réversibles. En revanche, l’utilisation adjuvante entraîne plus souvent des complications tardives irréversibles telles que lymphœdèmes, fibroses, raideurs articulaires, fractures de stress et sarcomes post-irradiation, de sorte que l’on cherche plutôt à les éviter.
Autres options thérapeutiques
En plus des traitements standard déjà mentionnés, il existe d’autres options thérapeutiques dont l’efficacité ou la supériorité n’ont pas encore été suffisamment démontrées par des études randomisées contrôlées. Il s’agit notamment, au niveau local, de l’hyperthermie régionale, de la perfusion hyperthermique isolée des extrémités, de la perfusion isolée des extrémités et de la curiethérapie locale par cathéter, et, au niveau systémique, des anticorps monoclonaux, des bisphosphonates, de la thérapie par cellules souches et de la nanotechnologie (photodynamique) [10].
Contrôles de suivi
Outre le suivi clinique initial, qui varie en fonction de l’intervention, le premier suivi clinico-radiologique, y compris la radiographie, a lieu trois mois après l’opération. localement une radiographie conventionnelle et une IRM de la zone tumorale, ainsi qu’un scanner thoracique pour exclure la présence de métastases (voir aussi www.sarcoma.ch). Comme la plupart des récidives ou des métastases tardives surviennent au cours des deux ou trois premières années, les contrôles de l’évolution sont effectués tous les trois mois à ce moment-là, avant que des consultations semestrielles ne soient organisées jusqu’au contrôle à cinq ans. Ensuite, le schéma sera adapté pour des contrôles sur un ou deux ans.
Conflit d’intérêt : les auteurs déclarent qu’il n’y a pas de conflit d’intérêt.
Littérature :
- HaDuong JH, et al : Sarcomas. Pediatr Clin North Am 2015 ; 62 : 179-200
- Miller MD, et al. Review of Orthopaedics. 6e édition. Philadelphie, PA : Elsevier Saunders 2012 ; 623-674.
- Kager L, et al : Ostéosarcome primaire métastatique : présentation et résultats des patients traités par des protocoles néoadjuvants du Groupe d’étude coopératif sur l’ostéosarcome. J Clin Oncol 2003 ; 21 : 2011-2018.
- Ferrone ML, Raut CP : Traitement chirurgical moderne : sauvetage du membre et rôle de l’amputation pour les sarcomes des tissus mous de l’extrémité. Surg Oncol Clin N Am 2012 ; 21 : 201-213.
- Jentzsch T, et al : Expression de MSH2 et MSH6 sur un microarray tissulaire chez des patients atteints d’ostéosarcome. Anticancer Res 2014 ; 34 : 6961-6972.
- Moore DD, Luu HH : Ostéosarcome. Cancer Treat Res 2014 ; 162 : 65-92.
- Grimer RJ, et al : Surgical outcomes in osteosarcoma. J Bone Joint Surg Br 2002 ; 84 : 395-400.
- Whelan JS, et al : EURAMOS-1, une étude internationale randomisée sur l’ostéosarcome : résultats du traitement pré-randomisé. Ann Oncol 2015 Feb ; 26(2) : 407-414.
- Yang JC, et al : Étude prospective randomisée du bénéfice de la radiothérapie adjuvante dans le traitement des sarcomes des tissus mous de l’extrémité. J Clin Oncol 1998 ; 16 : 197-203.
- Wunder JS, et al : Opportunités d’amélioration du ratio thérapeutique pour les patients atteints de sarcoma. Lancet Oncol 2007 ; 8 : 513-524
InFo ONKOLOGIE & HÄMATOLOGIE 2015 ; 3(5) : 18-21