Les termes de pneumopathie interstitielle ou de maladie parenchymateuse pulmonaire diffuse désignent un groupe de pathologies qui affectent l’épithélium des alvéoles pulmonaires (épithélium alvéolaire), l’endothélium des capillaires pulmonaires, la membrane basale ou les tissus périvasculaires et périlymphatiques des poumons. Dans la plupart des DLI, une fibrose peut se développer au cours de l’évolution.
Les termes maladie pulmonaire interstitielle (en anglais : Interstitial Lung Disease [ILD]) ou maladie pulmonaire parenchymateuse diffuse (en anglais : Diffuse Parenchymal Lung Disease [DPLD]) désignent un groupe de pathologies qui affectent l’épithélium des alvéoles pulmonaires (épithélium alvéolaire), l’endothélium des capillaires pulmonaires, la membrane basale ou les tissus périvasculaires et périlymphatiques des poumons. Souvent, les bronchioles et les bronches sont également touchées. Les ILD ne comprennent pas, par exemple, les maladies respiratoires obstructives ou les pneumonies directement liées à des agents pathogènes. Dans la plupart des DLI, une fibrose peut se développer au cours de l’évolution.
Répartition
Le groupe des ILD est hétérogène et peut être subdivisé de différentes manières. Le plus simple est de distinguer les pneumonies interstitielles dites idiopathiques (PII) d’une part, et les PII dont la cause est connue d’autre part. La cause d’une ILD peut être par exemple une maladie systémique comme la sarcoïdose ou les collagénoses, ainsi que des influences extérieures comme dans l’alvéolite allergique exogène (AAE), les pneumoconioses de la toxicité médicamenteuse ou la pneumonie radiogénique.
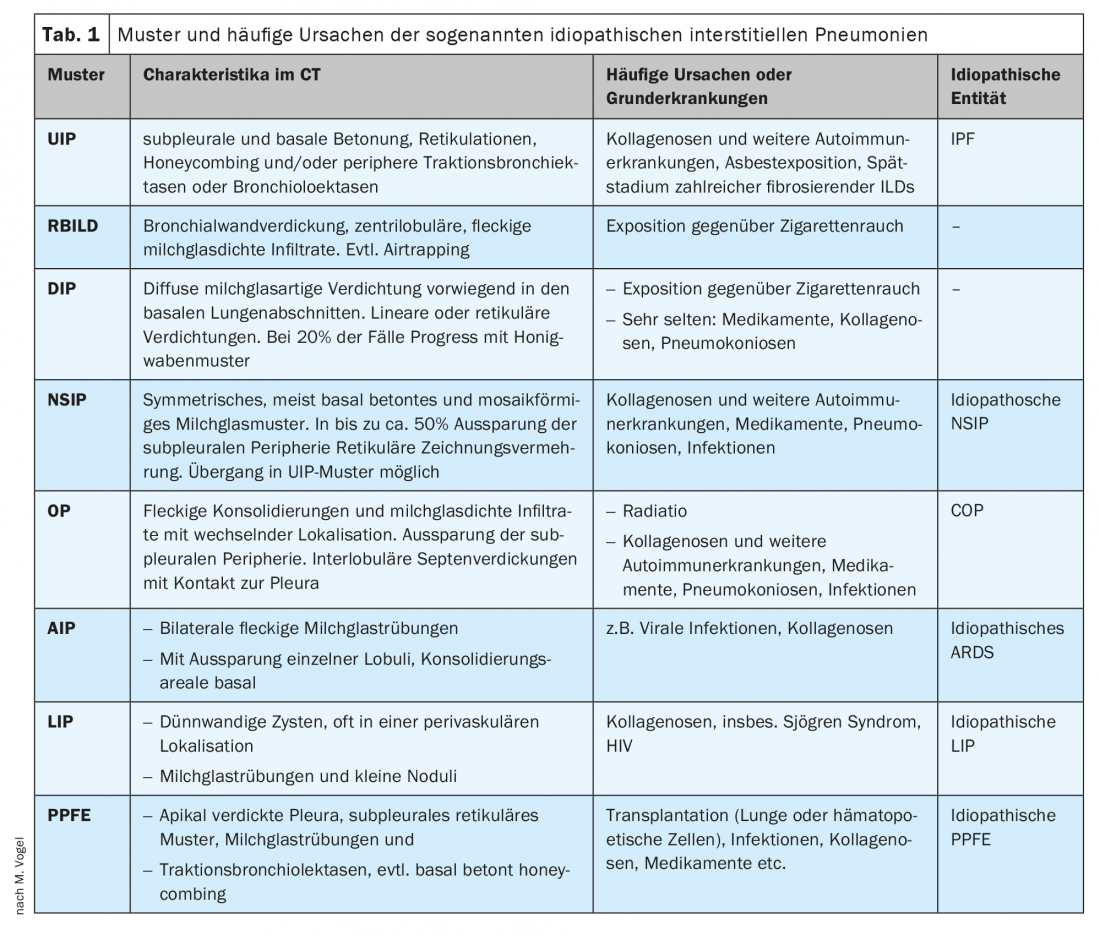
Pour les IIP, le scanner en couche mince et la préparation histologique distinguent principalement huit motifs et entités associées :
- Le modèle de pneumonie interstitielle (PI) ordinaire (“usual”) dans la fibrose pulmonaire idiopathique (FPI).
- Le schéma de bronchiolite respiratoire (BR) dans la pneumopathie interstitielle associée à la bronchiolite respiratoire (PIAR).
- Le schéma de la pneumonie interstitielle desquamative (PID) dans la PID du même nom.
- Le schéma NSIP dans la pneumonie interstitielle non spécifique (NSIP).
- Le modèle de pneumonie organisatrice (PO) dans la pneumonie organisatrice cryptogénique (POC).
- Le schéma de la pneumonie interstitielle aiguë (PIA) avec le tableau clinique du SDRA.
- Le schéma LIP dans la pneumonie interstitielle lymphoïde (PIL).
- Le schéma de la fibroélastose pleuroparenchymateuse dans la maladie du même nom (PPFE).
Ces schémas ne sont pas synonymes de maladie. Au contraire, ils peuvent d’une part être déclenchés de manière idiopathique ou d’autre part, il peut y avoir différentes causes connues : La fumée de cigarette pour RBILD et DIP et, entre autres, les collagénoses pour UIP, NSIP, OP, LIP et AIP [1] ou les pneumonies virales pour AIP, NSIP et OP (voir le tableau 1 avec les modèles et les causes fréquentes). Il n’existe pas de relation claire entre le motif et la maladie qui le déclenche : une même maladie peut déclencher différents motifs chez différents patients. De plus, les motifs présentent un spectre différent, avec parfois de grandes variations et des points communs avec d’autres motifs. L’étalon-or pour le diagnostic des ILD est donc le consensus interdisciplinaire d’au moins la pneumologie, la radiologie et la pathologie [2,3]. D’autres disciplines spécialisées, comme la rhumatologie, doivent également être régulièrement consultées. Dans ce contexte, bien que le schéma du scanner en couche mince soit généralement indicatif, le présent article montrera l’importance de toutes les disciplines spécialisées concernées et, en particulier, des informations anamnestiques et cliniques recueillies tout au long de l’évolution de la maladie pour l’établissement du diagnostic. L’accent sera mis ici sur la classification du modèle UIP.
Motifs UIP et IPF
La FPI est la plus fréquente et, avec une durée de survie médiane de 3 à 4 ans, la PII la moins favorable en termes de pronostic. La prévalence est de 2 à 29 pour 100 000 et l’incidence d’environ 10 pour 100 000/an [4]. Un traitement par des inhibiteurs spécifiques des fibroblastes peut retarder la progression, mais ne peut pas inverser la maladie [4]. Un traitement anti-inflammatoire, indiqué dans de nombreux autres IIP, a un impact négatif sur le pronostic [5]. Le modèle UIP présent dans la FPI peut également être non idiopathique, par exemple dans le cadre de collagénoses, d’une EAA fibrosante [6], après une exposition à l’amiante ou une sarcoïdose.
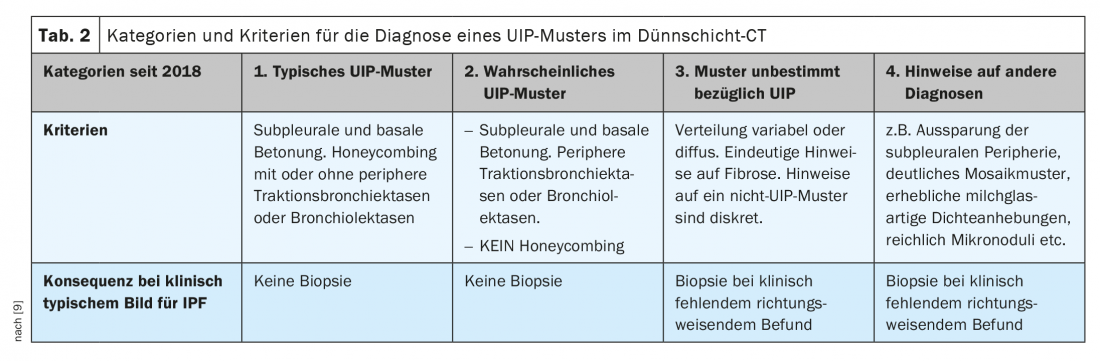
Une constellation typique de la FPI est la suivante : Sexe masculin, âge supérieur à 60 ans et antécédents de tabagisme, qui peuvent remonter à plusieurs années ou, à défaut, une incidence familiale accrue de FPI [3,7]. Si aucune maladie sous-jacente ni aucun effet n’est identifié chez un patient présentant une UIP, le diagnostic de FPI est posé. Il s’agit d’une maladie à part entière, qui peut être traitée, et dont l’origine semble multifactorielle. Le “schéma typique de l’UIP” (catégorie 1) se caractérise sur le plan morphologique du scanner par l’accentuation sous-pleurale et basale des modifications fibrotiques. On observe alors des réticulations et une structure pulmonaire en nid d’abeille, appelée honeycombing, avec ou sans bronchiectasies périphériques par traction ou bronchioloectasies. L’asymétrie est présente dans environ 25% des cas [8].
Les changements morphologiques typiques n’apparaissent pas dans tous les cas de PIF : Si seul le motif en rayon de miel est absent, on parle d’un “motif UIP probable” (catégorie 2). De plus, les informations cliniques ne sont généralement pas disponibles dans leur intégralité lors de la lecture de l’examen scanner. La première étape consiste donc à déterminer la certitude ou la probabilité d’un motif UIP à partir des images tomodensitométriques reconstruites en couches minces. De nouveaux critères ont été définis à cet effet en 2018 (tableau 2) [9].
La conséquence de la classification dans l’une des catégories oriente la suite de la démarche diagnostique. Dans un contexte clinique caractéristique de la FPI, la biopsie pulmonaire n’est pas pratiquée pour les catégories 1 et 2. [3,7,9].
Catégorie 1 : modèle UIP typique
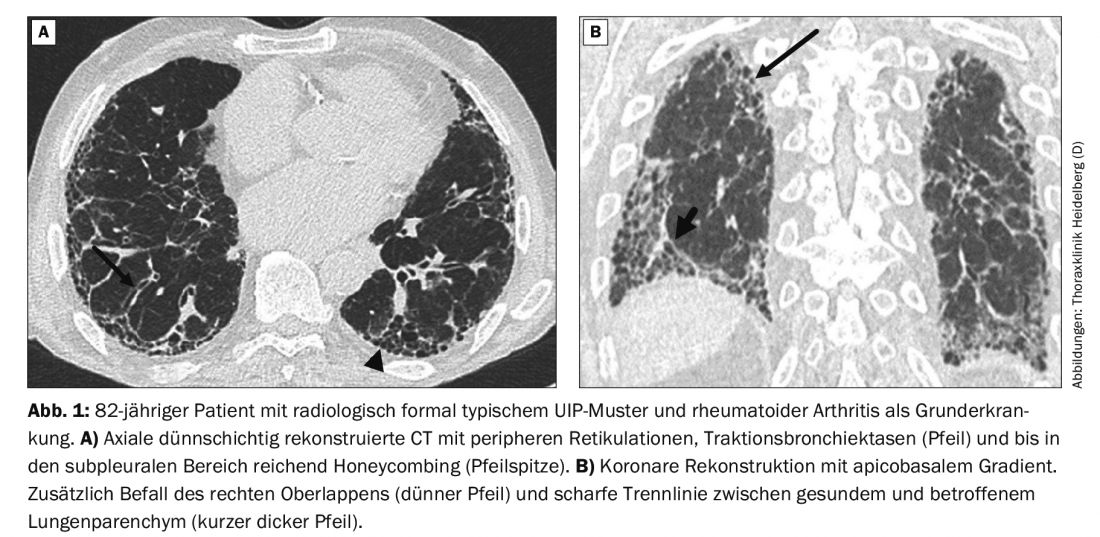
Un patient de 82 ans se présente avec une dyspnée d’effort progressive depuis 3-4 semaines. Il existe une toux sans expectoration. Il n’y a pas eu d’exposition à des substances dangereuses. L’anamnèse familiale est sans particularité. Jusqu’à il y a 45 ans, il y avait un abus occasionnel de nicotine avec un cumul de 4 à 5 paquets-années.
Le scanner natif réalisé par la suite avec des reconstructions en couches minces révèle un schéma typique de PIO (figure 1A). Lors de la réunion de concertation pluridisciplinaire, on apprend que le patient souffre également de polyarthrite rhumatoïde (PR). Par conséquent, le diagnostic posé n’est pas celui d’une FPI, mais celui d’une DLI associée à la PR.
Si un schéma de DLI survient dans le cadre d’une collagénose, le pronostic des patients est meilleur que dans le cas d’une FPI. Même en présence d’un schéma UIP formellement typique, probable ou indéterminé, il existe dans certains cas des indices morphologiques supplémentaires dans le parenchyme pulmonaire indiquant un lien avec une maladie auto-immune [10]. Dans le cas du patient de 82 ans, il s’agit de l’atteinte supplémentaire du lobe supérieur droit et de la ligne de démarcation oblique-horizontale nette entre le poumon sain et le poumon atteint (figure 1B). Ces signes ne sont toutefois pas probants et peuvent également être observés dans des cas de FPI.
Les données sur la fréquence de la pneumopathie interstitielle (PID) directement liée à la PR varient, car les symptômes respiratoires apparaissent souvent tardivement en raison de la limitation des mouvements. Une DLI cliniquement significative peut survenir chez environ 7% des patients atteints de PR [11]. Des indices radiologiques d’une DLI (généralement encore asymptomatique) peuvent toutefois être détectés sur des examens tomodensitométriques natifs reconstruits en coupes fines chez jusqu’à 60% de tous les patients atteints de PR [12]. L’abus de nicotine, même s’il remonte à plusieurs années, augmente également le risque de DLI associée à la collagénose et de type UIP [13].
Catégorie 2 : modèle UIP probable
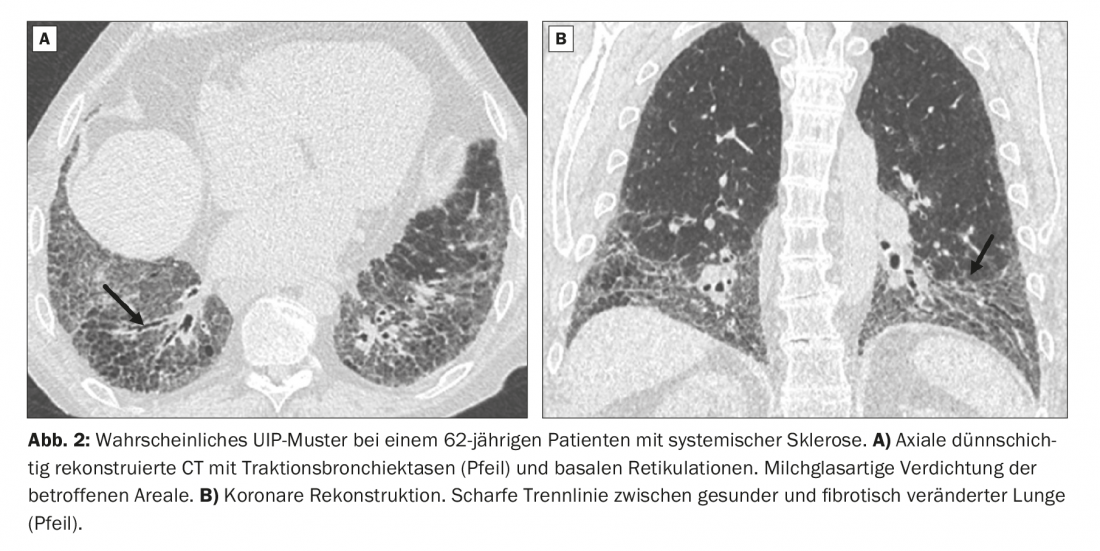
Un patient de 62 ans se présente avec une dyspnée d’effort lors d’un effort léger. Une sclérose systémique est déjà connue. Les images du scanner montrent un motif en bandes avec des bronchectasies et bronchiolectasies périphériques par traction avec une accentuation sous-pleurale et basale. De plus, une légère augmentation de la densité de type verre dépoli dans les zones concernées (Fig. 2A).
Du point de vue de la morphologie du scanner, l’image correspond formellement à celle d’un modèle probable d’UIP et devrait être classée comme telle sans connaissance de l’anamnèse. Mais dans ce cas, le diagnostic est celui d’une DLI dans le cadre d’une sclérose systémique. La ligne de démarcation horizontale nette entre le poumon sain et le poumon atteint est bien visible sur les images de scanner reformatées par coronarographie (figure 2B).
Catégorie 3 : modèle indéterminé par rapport à l’UIP ou au CT non indicatif d’un des diagnostics
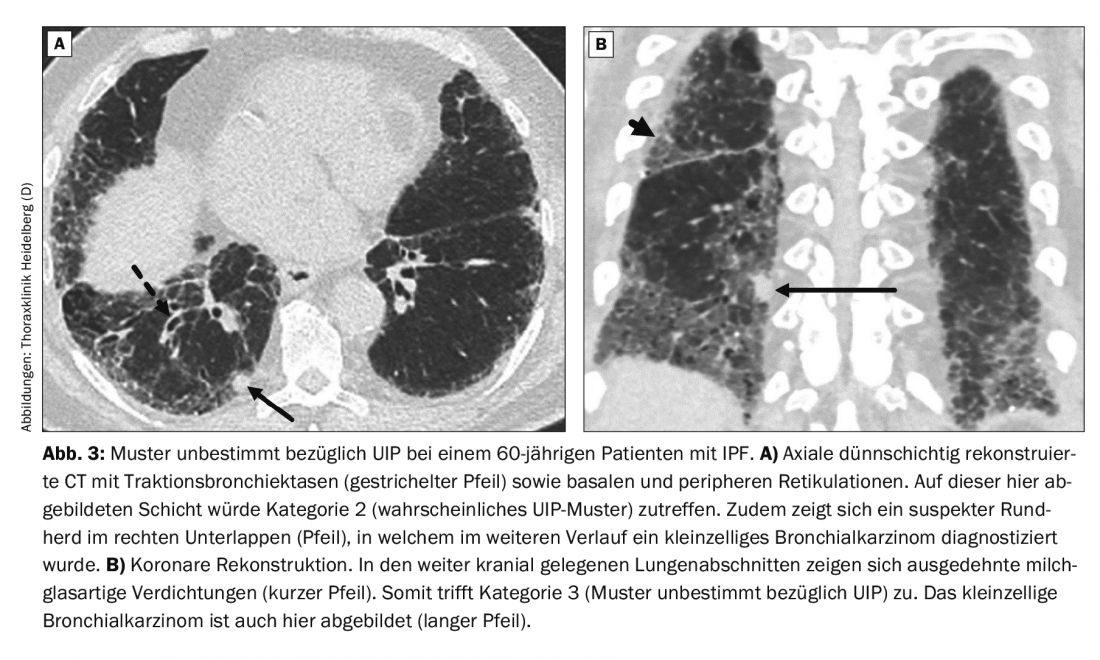
Un patient de 60 ans se présente avec une dyspnée. Jusqu’à il y a 10 ans, il y avait un abus de nicotine avec un total de 20 années de paquets. On ne connaît pas d’allergies, de maladies auto-immunes ou d’exposition à des substances dangereuses. Le scanner en coupes fines révèle un motif indéterminé par rapport à l’UIP (Fig. 3). L’examen histologique d’une biopsie chirurgicale du lobe inférieur droit a révélé un profil de PIO. Une FPI a été diagnostiquée. Un cancer bronchique à petites cellules a été découvert en complément.
Catégorie 4 : indices morphologiques d’un diagnostic non-IPF
Un patient de 71 ans atteint d’une cardiomyopathie dilatée et traité par amiodarone depuis 5 ans se présente avec une dyspnée qui augmente lentement. La pléthysmographie corporelle a révélé une pneumopathie restrictive modérée. Le motif sur le scanner en coupes fines n’est pas compatible avec un motif UIP (Fig. 4). Par exemple, l’évidement de la périphérie sous-pleurale et les augmentations importantes de densité en verre dépoli, même en dehors des zones touchées par la fibrose, sont incompatibles avec un motif UIP. Il existe une exception à cette règle : en cas de pneumonie interstitielle due à un agent pathogène ou d’exacerbation aiguë d’une FPI, des infiltrats étendus de densité en verre dépoli, associés à des modifications fibrotiques préexistantes, constituent le tableau typique [14].
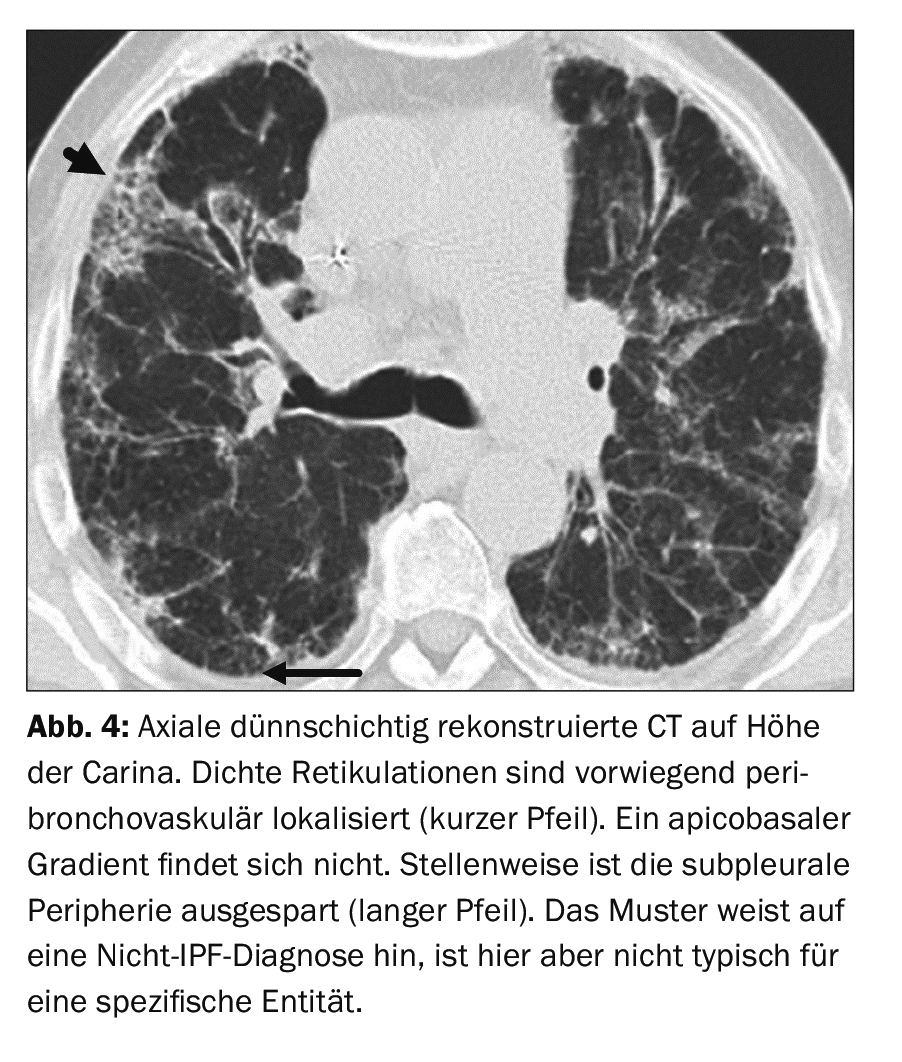
Une consolidation peut indiquer une opération. De même, une distribution principalement péribronchovasculaire, périlymphatique ou exclusivement dans les parties apicales ou moyennes des poumons indique un diagnostic de non-FPI. Un motif en mosaïque distinct, en particulier avec trois niveaux de densité différents et/ou des micronodules, sont des indices d’une EAA (en anglais hypersensitivity pneumonia, correspondant au terme de pneumonie d’hypersensibilité). D’ailleurs, une nouvelle directive a également été publiée en 2020 pour le diagnostic de l’EAA [6]. Dans le cas du patient de la figure 4 , une DLI d’origine médicamenteuse et toxique a été diagnostiquée. Cette maladie n’a pas de modèle spécifique ou pathognomonique.
Messages Take-Home
- Les pneumopathies interstitielles dites idiopathiques présentent des schémas caractéristiques en imagerie TDM.
- L’étalon-or pour le diagnostic des pneumopathies interstitielles est le consensus interdisciplinaire (médecine interne, radiologie, pathologie).
- Le modèle UIP est divisé en 4 catégories.
- La classification radiologique du motif UIP, associée aux données cliniques, a un impact direct sur la décision de réaliser ou non une biopsie pulmonaire supplémentaire et sur la décision thérapeutique.
Littérature :
- Capobianco J, et al : Manifestations thoraciques des maladies vasculaires du collagène. RadioGraphics 2012 ; 32 : 33-50.
- Travis WD, et al : An official American Thoracic Society/European Respiratory Society statement : Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013 ; 188 : 733-748.
- Lynch DA, et al : Critères de diagnostic de la fibrose pulmonaire idiopathique : un livre blanc de la Fleischner Society. The Lancet Respiratory medicine 2018 ; 6 : 138-153.
- Behr J : Diagnostic et possibilités de traitement de la fibrose pulmonaire idiopathique. Dtsch Arztebl International 2013 ; 110 : 875-881.
- Raghu G, et al : Prednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis. N Engl J Med 2012 ; 366 : 1968-1977.
- Raghu G, et al. : Diagnostic de la pneumonie d’hypersensibilité chez les adultes. Un guide de pratique clinique officiel ATS/JRS/ALAT. Am J Respir Crit Care Med 2020 ; 202 : e36-e69
- Brownell R, et al : The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017 ; 72 : 424-429.
- Tcherakian C, et al. : Progression de la fibrose pulmonaire idiopathique : leçons tirées de la maladie asymétrique. Thorax 2011 ; 66 : 226-231.
- Raghu G, et al. : Diagnostic de la fibrose pulmonaire idiopathique. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine 2018 ; 198 : e44-e68.
- Chung JH, et al : CT Features of the Usual Interstitial Pneumonia Pattern : Differentiating Connective Tissue Disease-Associated Interstitial Lung Disease from Idiopathic Pulmonary Fibrosis. American Journal of Roentgenology 2017 ; 210 : 307-313.
- Turesson C, et al : Manifestations de la maladie extra-articulaire dans l’arthrite rhumatoïde : tendances de l’incidence et facteurs de risque sur 46 ans. Annals of the rheumatic diseases 2003 ; 62 : 722-727.
- Brown KK : Maladie pulmonaire rhumatoïde. Actes de l’American Thoracic Society 2007 ; 4 : 443-448.
- Kumar A, et al : Concepts actuels de la pathogenèse, du diagnostic et de la prise en charge des maladies pulmonaires interstitielles liées au tabagisme. Chest 2018 ; 154 : 394-408.
- Akira M, et al : Résultats de tomographie computérisée dans l’exacerbation aiguë de la fibrose pulmonaire idiopathique. Am J Respir Crit Care Med 2008 ; 178 : 372-378.
InFo PNEUMOLOGIE & ALLERGOLOGIE 2021 ; 3(3) : 6-10