La destruction chronique des globules rouges caractérise l’hémoglobinurie paroxystique nocturne. Des mutations dans le gène PIG-A entraînent un déficit en GPI-AP, ce qui entraîne une activation incontrôlée du complément. Une inhibition du complément C5, qui ne doit être administrée que toutes les huit semaines pour la première fois, peut aider.
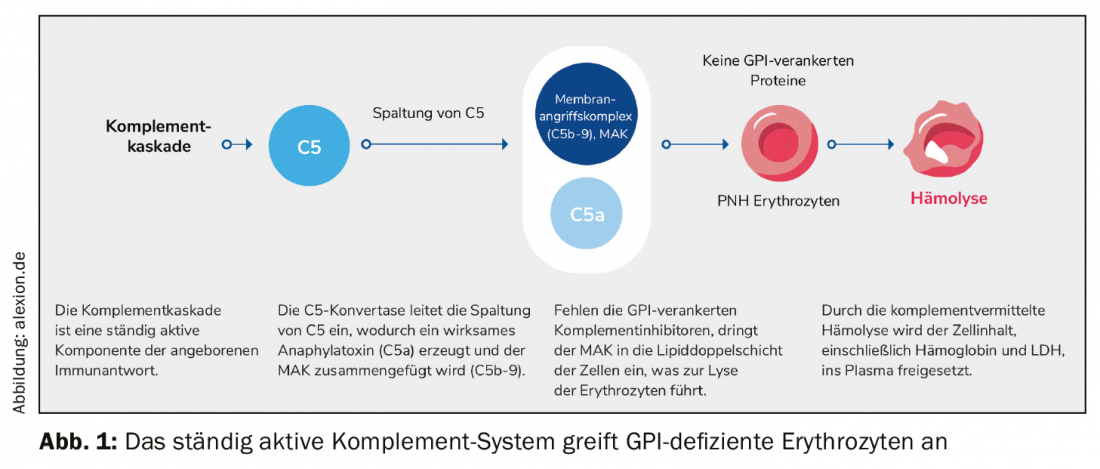
La principale caractéristique de l’hémoglobinurie paroxystique nocturne (HPN), une maladie rare, chronique et progressive, est une hémolyse médiée par le complément. En raison d’une mutation acquise, les cellules souches hématopoïétiques de la moelle osseuse sont dépourvues de protéines d’ancrage du glycophosphatidylinostiol (GPI). En conséquence, des protéines ancrées dans le GPI ne peuvent pas être formées, en particulier à la surface des érythrocytes rouges. La protection contre le système du complément, une partie du système immunitaire de l’organisme, disparaît. Les globules rouges sont pris pour des envahisseurs, attaqués et détruits. Le complexe d’attaque membranaire (CAM) pénètre dans la bicouche lipidique de la cellule et y déclenche la lyse des érythrocytes. Tout le contenu cellulaire, y compris l’hémoglobine et la LDH, est libéré dans le plasma (figure 1).
Maladie complexe avec des symptômes variables
La maladie se caractérise notamment par une anémie hémolytique, une hémoglobinurie et une insuffisance ou une défaillance de la moelle osseuse. Elle se manifeste classiquement par une fatigue, une diminution de la qualité de vie et une dyspnée d’effort. Mais des symptômes non spécifiques, souvent associés à des crises hémolytiques, peuvent également apparaître. Cependant, la cause principale de l’augmentation de la morbidité et de la mortalité des patients est la thrombophilie. Jusqu’à 50% des personnes touchées par la maladie et ne bénéficiant pas d’un traitement spécifique développent des thromboses. 35% des patients décèdent dans les 5 ans suivant le diagnostic.
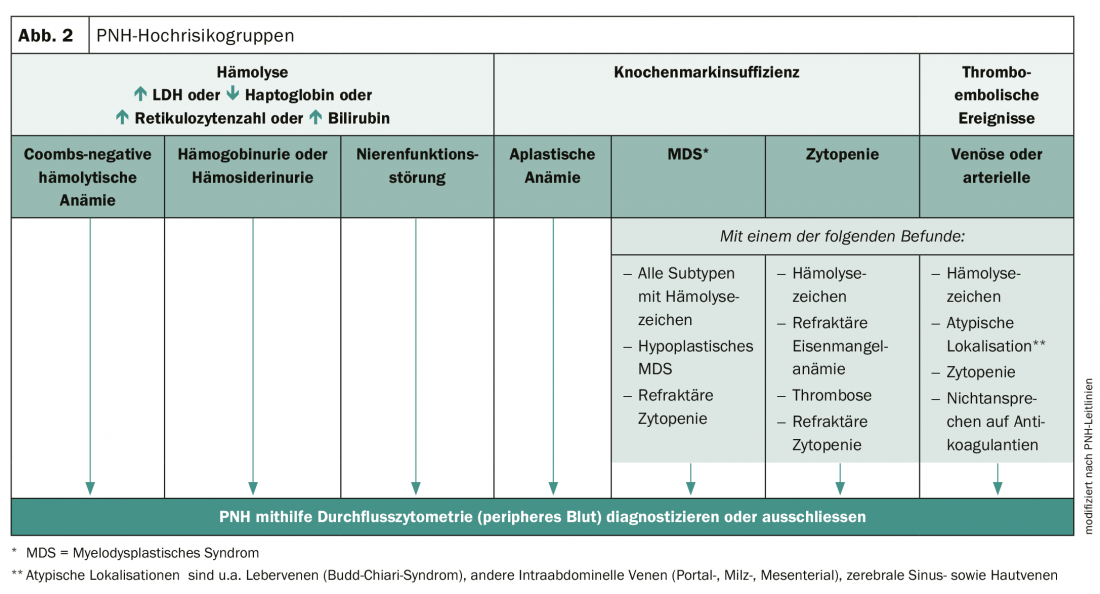
Un diagnostic précoce est essentiel pour améliorer le pronostic des personnes atteintes. L’HPN peut être diagnostiquée à l’aide de la cytométrie de flux à haute sensibilité et d’un examen clinique complet. Cependant, de nombreux médecins ne sont pas conscients de la complexité des symptômes et de la variabilité de la maladie. En conséquence, l’HPN n’est souvent pas détectée et le diagnostic n’est posé qu’avec un retard allant de 1 à plus de 5 ans. Il existe toutefois des groupes à haut risque chez lesquels la présence d’une HPN doit être envisagée (figure 2).
La gestion du traitement s’améliore
Pour stopper l’hémolyse, on intervient sur les fonctions terminales de la cascade du complément au moyen d’un inhibiteur du complément. En revanche, les fonctions proximales sont maintenues. Par exemple, le taux d’événements thromboemboliques (ET) a été significativement réduit de 11,54 à 0,72 chez les patients traités par anticoagulant. L’inhibiteur s’est également montré convaincant en termes de bénéfices cliniques à long terme. Seul bémol : l’intervalle de perfusion court. Aujourd’hui, l’inhibiteur a pu être développé et sa demi-vie prolongée (ravulizumab). La réduction à 6-7 perfusions par an améliore considérablement la qualité de vie des personnes concernées et a également permis de réduire le risque d’hémolyse de rupture de 10,7% à 4%.
Source : Réunion annuelle 2019 des sociétés savantes germanophones d’hématologie et d’oncologie médicale (DGHO)
InFo ONKOLOGIE & HÄMATOLOGIE 2019 ; 7(6) : 32-33 (publié le 6.12.19, ahead of print)