Le syndrome de Von Hippel-Lindau est dû à des mutations du gène VHL et se caractérise par des hémangioblastomes du cerveau, de la moelle épinière et de la rétine de l’œil. En outre, les personnes atteintes présentent un risque accru de carcinomes et de kystes rénaux, de phéochromocytomes, de kystes et de tumeurs neuroendocrines du pancréas, de tumeurs du sac endolymphatique et de kystes de l’épididyme, ainsi que des ovaires et des trompes de Fallope.
Les symptômes dépendent de la taille et de l’emplacement des tumeurs [1]. Les enfants peuvent souffrir de maux de tête et se sentir étourdis ou faibles. Des troubles de la vision, qui peuvent entraîner une perte de vision en cas de croissance de la tumeur rétinienne, et une hypertension peuvent également survenir. Il peut y avoir une perte de coordination. Environ 10% des enfants concernés ont une tumeur de l’oreille interne, ce qui peut affecter leur audition. Sans traitement, les personnes atteintes peuvent devenir aveugles, subir des lésions cérébrales ou mourir. Les décès sont généralement dus à des complications des angiomes cérébraux ou du cancer du rein.

Un diagnostic précoce est d’une importance capitale. Sur le plan phénotypique, on distingue différents types de syndrome de Von Hippel-Lindau [1] : Le type I est caractérisé par l’apparition d’hémangiomes dans la rétine et/ou le système nerveux central, de carcinomes des cellules rénales et/ou de tumeurs neuroendocrines. Le risque de phéochromocytome est toutefois très faible. Le type I est associé à des mutations non sens ou à des délétions de grandes sections de gènes. En revanche, le type II est souvent associé à des mutations missense et le risque de développer des phéochromocytomes est très élevé.
Critères de diagnostic
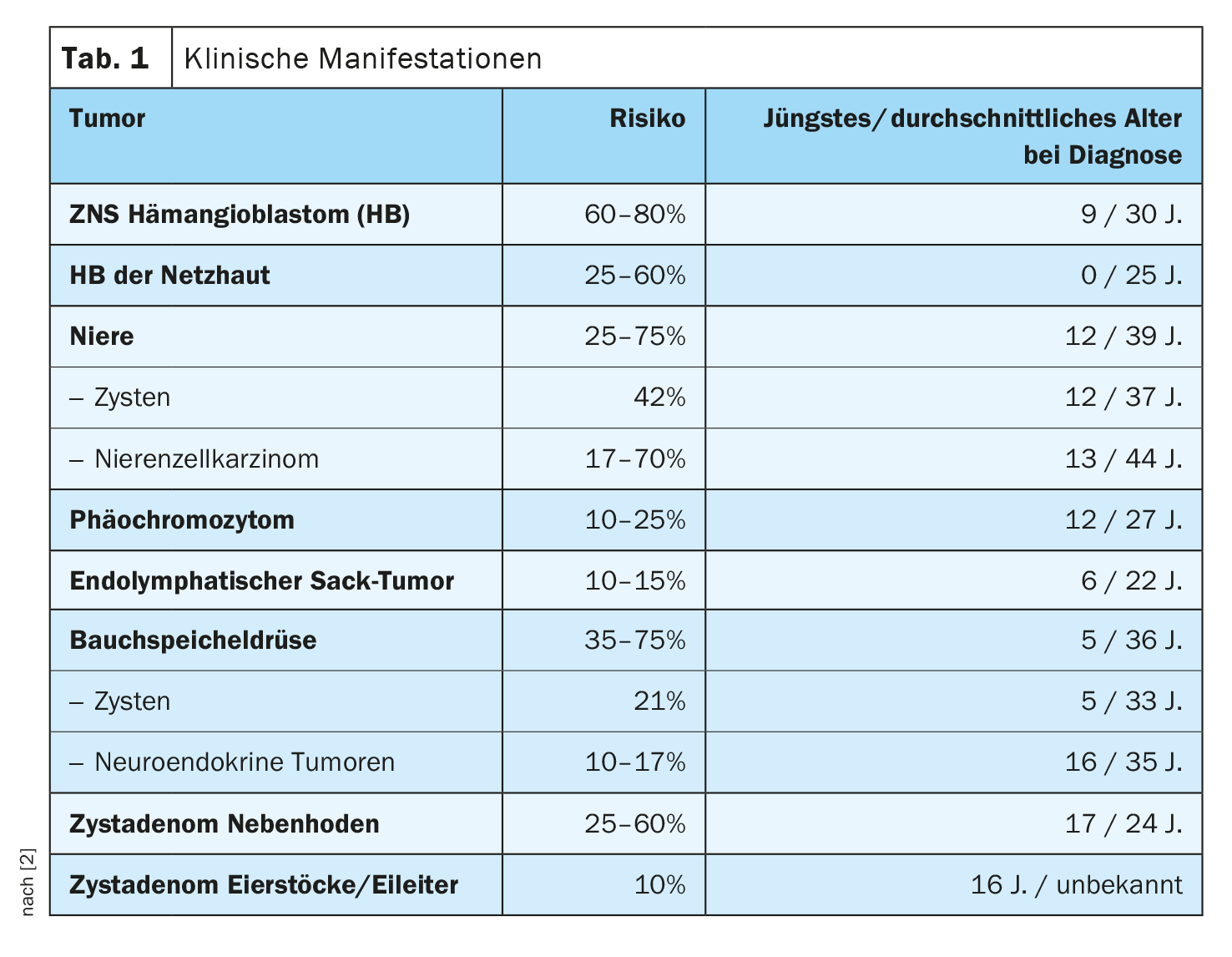
Le diagnostic de syndrome de Von Hippel-Lindau est considéré comme certain lorsqu’une mutation est détectée dans le gène VHL et/ou [2] :
sans syndrome de Von Hippel-Lindau dans la famille en présence d’au moins 2 des constats suivants :
≥2 hémangioblastomes de la rétine, de la moelle épinière ou du cerveau, ou un hémangioblastome unique associé à une manifestation dans l’abdomen (par ex. plusieurs kystes des reins ou du pancréas)
– Carcinome des cellules rénales
– Phéochromocytome
– ELST, cystadénome des ovaires ou des trompes de Fallope/épididyme ou tumeurs neuroendocrines du pancréas
Avec syndrome de Von Hippel-Lindau dans la famille en présence d’au moins 1 des constats suivants :
– Hémangioblastome de la rétine
– Hémangioblastome de la moelle épinière ou du cervelet
– Phéochromocytome
– Carcinome des cellules rénales
– Plusieurs kystes des reins ou du pancréas
Littérature :
- Institut für Klinische Genetik, Universitätsklinikum Carl Gustav Carus Dresden, www.uniklinikum-dresden.de, dernière consultation 17.01.2023)
- Medizinische Hochschule Hannover, www.krebs-praedisposition.de/fuer-patienten-und-familien/von-hippel-lindau-syndrom/#diagnose, (dernière consultation 17.01.2023)
- Baumgartner-Parzer S: J Klin Endokrinol Stoffw 2020; 13: 37–40.
HAUSARZT PRAXIS 2023; 18(1): 47
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(1): 32