Les vascularites systémiques sont des syndromes inflammatoires des vaisseaux sanguins qui peuvent provoquer un très large éventail de symptômes en fonction du calibre des vaisseaux concernés et de leur localisation. Les vascularites primaires les plus fréquentes chez l’adulte sont décrites avec les options thérapeutiques médicamenteuses actuelles.
Les vascularites systémiques sont des syndromes inflammatoires des vaisseaux sanguins qui peuvent provoquer un très large éventail de symptômes en fonction du calibre des vaisseaux concernés et de leur localisation. Une atteinte vasculaire des petits vaisseaux, c’est-à-dire des capillaires, des artérioles et des veinules, peut par exemple se manifester, selon sa localisation, par un purpura palpable de la peau, une glomérulonéphrite nécrosante à progression rapide avec insuffisance rénale, des hémorragies pulmonaires, des saignements de nez, une sclérite ou une opacité cérébrale. Si les artères de moyen et grand calibre sont touchées, il existe un risque d’infarctus des tissus, d’anévrisme, d’hémorragie et de thrombose. Bien que des progrès significatifs aient été réalisés dans le traitement des vascularites systémiques au cours des 40 dernières années, la mortalité reste nettement plus élevée que dans la population générale [1]. Les vascularites primaires les plus fréquentes chez l’adulte sont décrites ci-dessous, avec les options thérapeutiques médicamenteuses actuelles.
Classification des vascularites systémiques
Le groupe des vascularites systémiques “primaires” comprend des syndromes pathologiques “idiopathiques” indépendants, tandis que les vascularites “secondaires” sont associées à des maladies préexistantes. La vascularite cryoglobulinémique associée à l’hépatite C et la vascularite associée à la polyarthrite rhumatoïde séropositive de longue date sont des exemples de vascularites secondaires.
La classification des vascularites systémiques primaires selon la classification de Chapel Hill est basée sur le calibre des vaisseaux atteints [2]. (Aperçu1). Les vascularites des gros vaisseaux, qui affectent l’aorte et/ou ses branches, comprennent l’artérite de Takayasu, qui touche surtout les jeunes femmes asiatiques, et l’artérite à cellules géantes (ACG ; synonyme : artérite temporale), qui ne se manifeste généralement pas avant l’âge de 50 ans. Le syndrome de Kawasaki, qui touche les enfants, et la très rare panartérite noueuse affectent particulièrement les artères de moyen calibre. Les vascularites primaires des petits vaisseaux se divisent en deux groupes principaux : Les vascularites associées aux ANCA (anti-neutrophil cytoplasmic antibodies) (AAV) et les vascularites ANCA-négatives. Les AAV comprennent la granulomatose avec polyangéite (GPA ; anciennement maladie de Wegener) ; la polyangéite microscopique (MPA) ; la granulomatose éosinophile avec polyangéite (EGPA ; anciennement syndrome de Churg-Strauss) ; et la glomérulonéphrite nécrosante focale (FNGN). Les vascularites primaires des petits vaisseaux ANCA-négatives sont associées à un certain nombre de vascularites à médiation par des complexes immuns, telles que la vascularite à IgA (Henoch-Schönlein), qui survient principalement chez les enfants, et la vascularite cryoglobulinémique essentielle.

Les vascularites secondaires affectent généralement les petits vaisseaux (capillaires, artérioles, veinules) et, comme les vascularites des petits vaisseaux ANCA-négatives, elles sont généralement causées par des complexes immuns avec activation du système du complément. Le traitement des vascularites secondaires se fait toujours en fonction de la maladie sous-jacente et implique très souvent, lorsque le rapport bénéfice/risque est donné, l’utilisation à court terme de glucocorticoïdes à plus forte dose, et dans certains cas (par exemple la vascularite cryoglobulinémique induite par un virus) la déplétion thérapeutique des cellules B CD20+ [3]. Parmi les syndromes vasculaires, la maladie de Behçet occupe une place un peu particulière, car elle peut toucher tous les calibres de vaisseaux.
Pour toutes les vascularites, le premier objectif du traitement est d’obtenir le plus rapidement possible une rémission clinique aussi complète que possible, ce que l’on appelle l’induction de la rémission. Vient ensuite la phase de maintien de la rémission, qui peut durer plusieurs années selon le type de vascularite.
Artérite à cellules géantes (ACG)
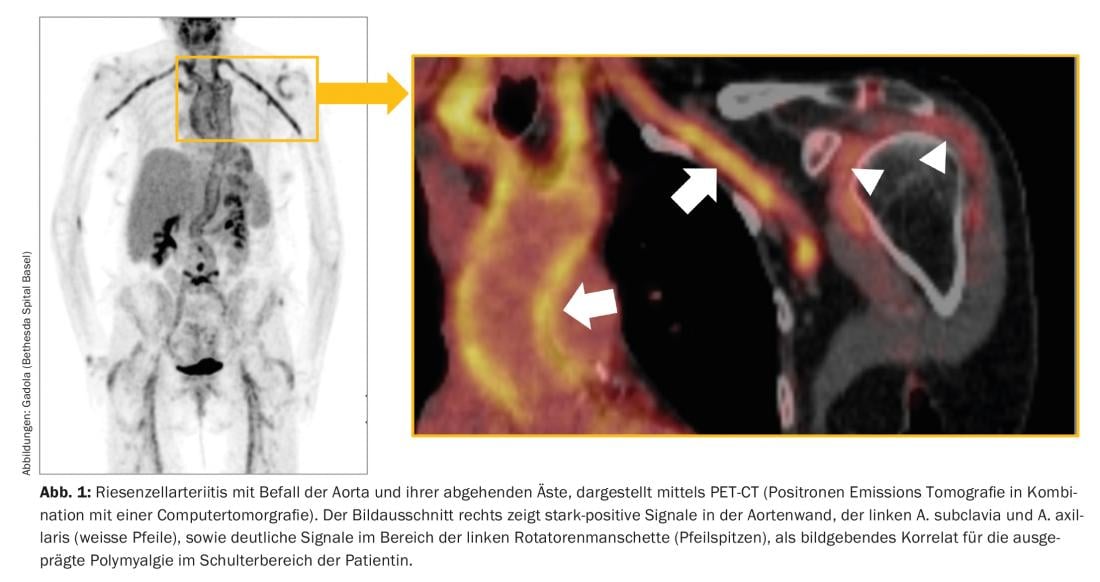
L’ACR, également connue sous le nom d’artérite temporale, est la vascularite la plus fréquente sous nos latitudes et apparaît souvent après l’âge de 60 ans, et dans la grande majorité des cas après l’âge de 50 ans. Elle affecte l’aorte et les artères de gros et moyen calibre qui en découlent (fig. 1). Les symptômes typiques sont des céphalées nouvelles, souvent “névralgiques”, un syndrome polymyalgique, une symptomatologie B avec baisse de performance, épuisement et sueurs nocturnes, des symptômes de claudication des extrémités, de la langue et/ou des muscles masticateurs, ainsi que des troubles visuels. La RZA est surtout redoutée en raison du risque de cécité aiguë, qui est généralement irréversible. En laboratoire, une réaction prononcée en phase aiguë est typique – mais pas obligatoire -, avec une nette accélération de la réaction de sédimentation (BSR) et une augmentation de la protéine C-réactive (CRP).
Les glucocorticoïdes sont les médicaments de premier choix pour le RZA et entraînent typiquement une amélioration spectaculaire des symptômes subjectifs dans les 24 heures. En cas de suspicion d’atteinte oculaire, par exemple en cas de troubles passagers de l’acuité visuelle, et de menace potentielle de cécité, de fortes doses sont administrées par voie intraveineuse, par exemple 1 g de méthylprednisolone pendant trois jours consécutifs. Dans d’autres cas, une dose quotidienne initiale de 40-60 mg est suffisante. La dose est ajustée en fonction de la clinique et des résultats de laboratoire. Tant que la dose de prednisone ne peut pas être réduite en dessous de 20 mg/jour, une antibioprophylaxie par sulfométoxazole-trimétoprime (p. ex. Cotrim-CT 800/160 mg ; Bactrim forte®) doit être administrée 3×1/semaine pour éviter les infections opportunistes, notamment celles dues à Pneumocystis jirovecii.
Auparavant, dans le cas du RZA, comme dans d’autres vascularites, des doses de prednisone de 1 mg/kg de poids corporel étaient utilisées pendant toute une année, avec une survenue fréquente d’effets secondaires graves. Cependant, grâce aux médicaments antirhumatismaux de base (DMARD), qui permettent d’économiser les stéroïdes, la dose de prednisone peut être réduite bien plus tôt en dessous de la dose seuil de stéroïdes de 7,5 mg de prednisone. Le méthotrexate (MTX), en particulier, a fait ses preuves en tant que DMARD pour le RZA. Le début de l’effet du MTX, comme celui de tous les DMARD, est retardé et se produit environ 4 à 6 semaines après que la dose efficace a été atteinte pour le MTX.
Le méthotrexate doit toujours être administré par voie parentérale, c’est-à-dire par voie sous-cutanée (s.c.) une fois par semaine, pour le traitement des vascularites. En cas de bonne tolérance, la dose peut être augmentée jusqu’à un maximum de 0,3 mg/kgKG par semaine. La BSR et la CRP servent également de biomarqueurs évolutifs précieux de l’activité inflammatoire sous traitement par la prednisone et les DMARD. Pour réduire les effets secondaires toxiques du MTX, il faut toujours prendre de l’acide folique en accompagnement, par exemple 10 mg/semaine, le jour suivant l’injection de méthotrexate. Le MTX ne doit jamais être associé au sulfométoxazole-trimétoprime, sous peine d’entraîner une myélosuppression sévère. C’est pourquoi nous n’utilisons le MTX qu’après avoir réduit la dose de prednisone à moins de 20 mg/jour.
Avant de commencer un traitement de fond d’épargne stéroïdienne avec du MTX ou d’autres DMARD, il convient de réaliser une radiographie du thorax pour exclure une infection chronique ou une fibrose pulmonaire, une numération formule sanguine différenciée, des tests hépatiques et rénaux, ainsi que des tests sérologiques pour l’hépatite B, l’hépatite C et le VIH. Pendant les 3 premiers mois sous méthotrexate, il est recommandé de contrôler les valeurs hépatiques et rénales ainsi que la formule sanguine tous les mois ; par la suite, ces contrôles peuvent éventuellement être espacés de 8 à 12 semaines. Des recommandations détaillées sur l’utilisation des DMARD dans les maladies rhumatologiques sont disponibles sur les portails Internet des sociétés de rhumatologie [par ex. 4].
Depuis 2017 (espace UE) resp. 2018 (Suisse), l’anticorps anti-récepteur de l’interleukine 6 (anti-IL6R) tocilizumab (Actemra®) a été approuvé pour le traitement du RZA. Le tocilizumab permet de réduire beaucoup plus rapidement la dose de glucocorticoïdes, même sans méthotrexate, avec un bon résultat clinique [5,6]. Le traitement par tocilizumab doit être poursuivi pendant au moins 1 an, sinon les récidives sont fréquentes [7]. La BSR et la CRP sont supprimées sous tocilizumab et ne sont donc pas utiles comme paramètres d’évolution de l’activité de la maladie. Des données récentes suggèrent que la dose de glucocorticoïdes sous tocilizumab pourrait être arrêtée très rapidement, en l’espace de quelques semaines [8] ; toutefois, des études supplémentaires sont nécessaires avant de pouvoir formuler des recommandations claires. Étant donné que les glucocorticoïdes ne peuvent actuellement pas être réduits en dessous de la dose seuil pendant une période prolongée (>3 mois) en cas de RZA, il est recommandé de commencer rapidement un traitement antirésorptif, par exemple avec de l’alendronate, afin d’éviter une résorption osseuse accélérée par les stéroïdes ou une perte osseuse. d’une ostéoporose.
Artérite de Takayasu (TA)
Comme le RZA, l’AT affecte l’aorte et les grosses artères qui en partent. Contrairement au RZA, cette vascularite survient dans 80 à 90% des cas chez les femmes, avec un début de la maladie entre 10 et 40 ans . L’AT évolue par poussées et se manifeste typiquement par des symptômes constitutionnels, des arthralgies et, de manière caractéristique, une douleur marquée à la pression des carotides (dans 10 à 30% des cas). Au cours de la maladie, des occlusions vasculaires, une hypertension rénovasculaire sévère, une rétinopathie (de Takayasu), ainsi que des anévrismes aortiques avec ou sans insuffisance valvulaire aortique peuvent survenir.
Si un TA est diagnostiqué, les glucocorticoïdes sont utilisés en premier lieu. Les DMARDs modificateurs de la maladie économes en stéroïdes les plus utilisés dans l’AT sont le méthotrexate s.c. (comme pour le RZA) ou l’azathioprine p.o. (jusqu’à 2 mg/kgKG). Les alternatives sont le mycophénolate (1,5 g-3 g/jour p.o.) et le léflunomide (20 mg/jour p.o.). En cas d’intolérance au méthotrexate ou aux DMARD oraux, de cas résistants au traitement et de cas graves, on utilise des bloqueurs du TNFalpha- (p. ex. étanercept ou infliximab) ou d’autres produits biologiques (p. ex. tocilizumab, abatacept, ustékinu-mab), qui ne sont toutefois pas encore autorisés pour cette indication [9].
Vascularites associées aux ANCA (AAV)
GPA (anciennement maladie de Wegener)
L’AMP est la vascularite primitive des petits vaisseaux la plus fréquente sous nos latitudes, avec une répartition presque équilibrée entre les sexes et un âge typique de manifestation entre 40 et 60 ans. l’âge de 18 ans. On distingue une “phase initiale” granulomateuse non vascularisée (en anglais “localized”) et une “phase de généralisation” vascularisée systémique, qui peuvent se produire de manière séquentielle (phase initiale ‘ phase de généralisation) ou simultanée. Alors qu’il existe aujourd’hui un large éventail de traitements efficaces pour la vascularite des petits vaisseaux, le traitement des manifestations inflammatoires granulomateuses agressives représente souvent un grand défi [10,11].
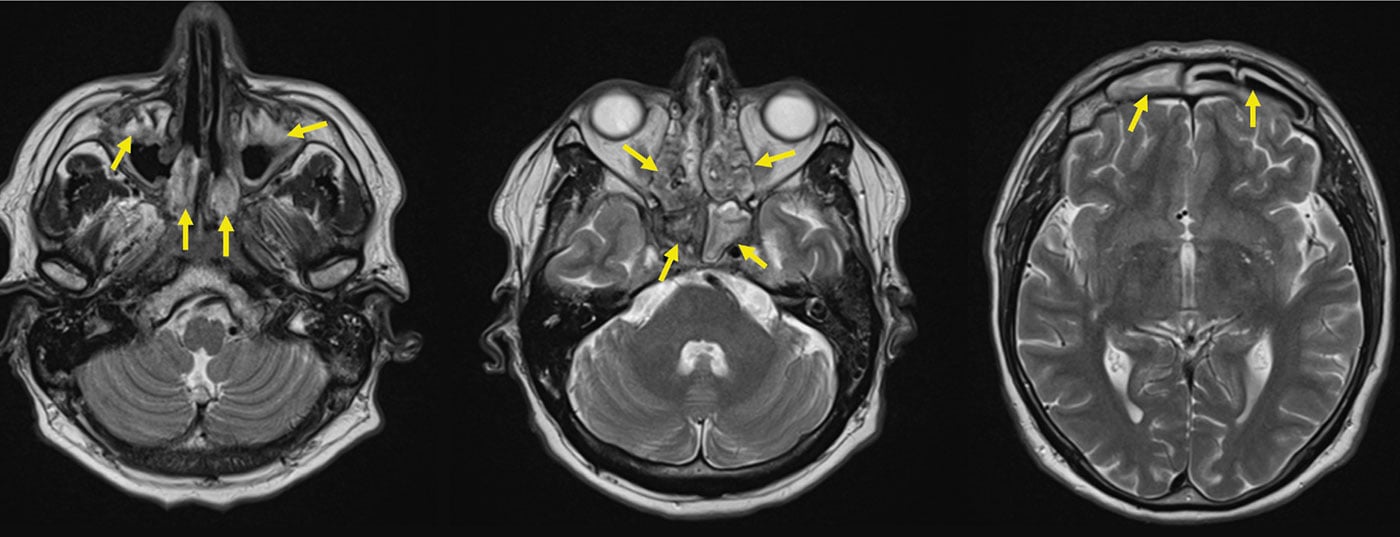
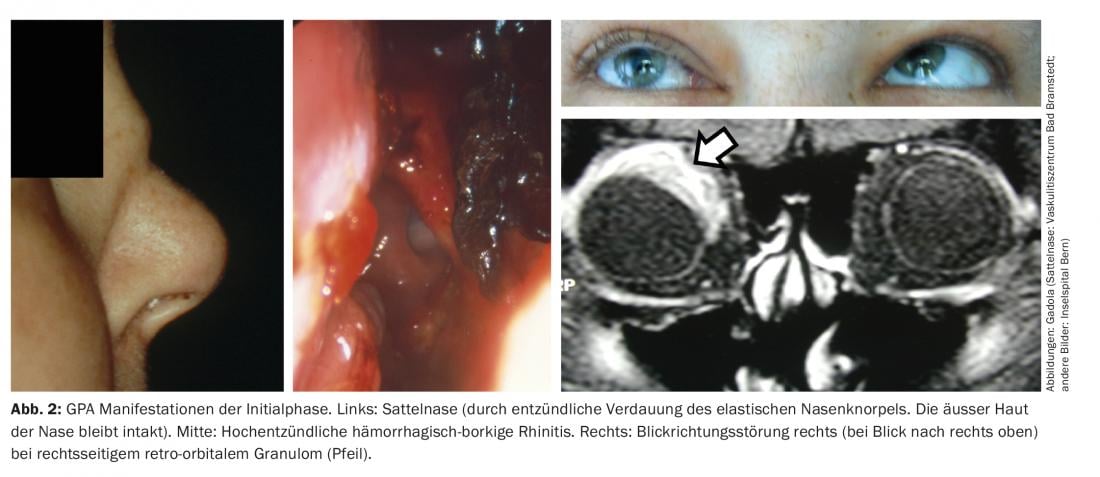
Phase initiale de l’AMP (AMP “localisée”). La phase initiale de l’AMP se manifeste typiquement au niveau de la gorge, du nez, des oreilles et des voies respiratoires, par exemple sous la forme d’une rhinite chronique hémorragique et boréale, d’une sinusite résistante au traitement ou d’une mastoïdite. En cas d’évolution agressive, on peut observer un nez en selle, par destruction du cartilage nasal élastique, des fistules dans l’orbite ou vers le visage, des granulomes rétro-orbitaires avec troubles de la direction du regard (diplopie), et une pachyméningite granulomateuse (fig. 2). Ces deux dernières sont probablement dues à une inflammation granulomateuse per continuitatem provenant des sinus [11]. Les ANCA spécifiques de la protéinase-3 (PR3-ANCA) ou, plus rarement, de la myéloperoxydase (MPO-ANCA) ne sont détectables que chez environ 50% des patients atteints d’AMP pendant la phase initiale.
Pour traiter la phase initiale “pure” sans vascularite systémique associée, différentes combinaisons de médicaments sont utilisées en fonction de la sévérité des symptômes. Dans les cas moins graves, le sulfométhoxazole-trimétoprime (T/S) est utilisé avec ou sans prednisone à faible dose (10 mg/jour). L’utilisation de la T/S remonte à une observation empirique faite dans les années 1970 par Richard Deremee à la Mayo Clinic [12]. Dans les années 1990, une association entre la colonisation nasale chronique par Staphylococcus aureus et l’activité de la maladie dans l’AMP a été démontrée [13]. Depuis lors, le traitement antibiotique topique intra-nasal à la mupirocine est également utilisé, mais sans succès éclatant. Une analyse récente du microbiome endonasal dans l’AMP a montré un lien intéressant entre l’activité de la maladie dans l’AMP et le Corynebacterium tuberculostearicum [14]. Cet agent pathogène est un pathogène important dans d’autres maladies granulomateuses [15]. Corynebacterium tuberculostearicum est résistant à la plupart des antibiotiques [16], ce qui pourrait expliquer le succès modéré de la T/S et de la mupirocine.
En cas d’évolution plus agressive de la phase initiale, on utilise en premier lieu le méthotrexate (toujours sans T/S, sinon toxicité combinée de la moelle osseuse) en association avec la prednisone. Dans les cas réfractaires, les grands granulomes pulmonaires et aussi les granulomes rétro-orbitaires, le traitement par anticorps anti-CD20 (rituximab) peut être efficace [17]. Les granulomes dans l’AMP présentent une forte densité de cellules B CD20+ et sont considérés comme un site d’origine possible de la production d’ANCA [18,19]. Une manifestation particulière de l’AMP pendant la phase initiale est la trachéotomie inflammatoire et fibrosante avec dyspnée et stridor inspiratoire, qui peut nécessiter un traitement par infiltration locale de glucocorticoïdes et dilatation par ballonnet.
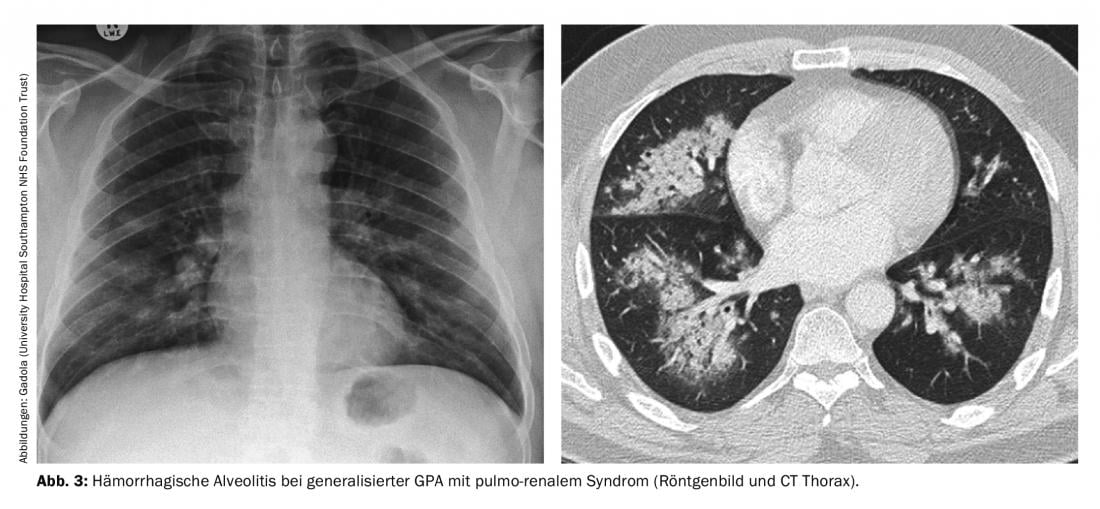
Phase de généralisation de la GPA (ANCA dans >98%). La vascularite systémique des petits vaisseaux dans l’AMP, dans laquelle des PR3-ANCA ou des MPO-ANCA sont pratiquement toujours détectés dans le sérum, peut toucher tous les organes. L’atteinte des reins entraîne typiquement une glomérulonéphrite pauci-immune nécrosante à progression rapide (RPGN) qui, non traitée, peut rapidement conduire à une insuffisance rénale sévère nécessitant une dialyse. Le syndrome pulmo-rénal est particulièrement redouté. la survenue combinée d’une RPGN et d’une alvéolite hémorragique (figure 3), qui présente une mortalité élevée. Les autres manifestations typiques de la phase de généralisation sont une symptomatologie B évidente, des sclérites – la sclérite nodulaire douloureuse est typique – qui peuvent conduire à une scléromalacie, une mononévrite multiplex souvent très douloureuse, une encéphalite avec opacification, un purpura palpable, et une polyarthrite non érosive. Les principes de traitement suivants pour l’AMP généralisée peuvent être appliqués à la MPA et à la FNGN, et en partie à l’EGPA.
Induction de la rémission dans l’AMP généralisée. Différents principes actifs sont disponibles pour induire une rémission, en fonction de la sévérité des manifestations. En premier lieu, l’activité inflammatoire de l’AMP généralisée doit toujours être contrôlée le plus rapidement et le plus soigneusement possible.
En cas d’AMP systémique non rénale et ne menaçant pas d’autres organes, le traitement d’induction de la rémission peut être initié avec du méthotrexate plus de la prednisone ou avec des pulsations intraveineuses (i.v.) de cyclophosphamide (par ex. 4 pulsations de 10 mg/kgKG à 3 semaines d’intervalle) plus de la prednisone, à condition que des contrôles cliniques étroits, au moins une fois par semaine, soient possibles. En cas de manifestations rénales et d’autres manifestations menaçant les organes, on utilise en premier lieu, outre la prednisone, le cyclophosphamide par voie orale (en commençant par 2 mg/kgKG/jour) ou sous forme de pulsation i.v., ou le rituximab (2 perfusions i.v. de 1g de rituximab à 14 jours d’intervalle) en combinaison avec la prednisone. Le choix entre le traitement par cyclophosphamide oral, plus intensif en termes de doses, et le traitement par impulsions nécessite toujours une évaluation minutieuse du rapport bénéfice/risque par un spécialiste expérimenté dans ce domaine. Sous cyclophosphamide, les leucocytes en particulier, et plus particulièrement les granulocytes neutrophiles, qui sont typiquement nettement plus élevés en cas de vascularite active à AMP, doivent être régulièrement déterminés. Sous cyclophosphamide, les leucocytes devraient atteindre le nadir en 8 à 10 jours, lequel devrait toujours être déterminé. Si les leucocytes ne chutent pas, cela indique une activité persistante de la maladie, ce qui peut nécessiter d’augmenter la dose “adaptée aux leucocytes” à 2,5-3 mg/kgKG. Sous cyclophosphamide, des infections opportunistes graves peuvent survenir peu de temps après le début du traitement, en cas d’apparition d’une leucopénie, notamment la réactivation du cytomégalovirus (CMV). Les effets secondaires toxiques typiques liés à la dose dans la vessie sont la cystite hémorragique et le carcinome de la vessie, qui surviennent après une dose cumulée d’au moins 25 g (et en moyenne de 100 g) de cyclophosphamide [20], ainsi que le syndrome myélodysplasique (SMD). La probabilité de complications vésicales peut être réduite par une bonne hydratation pendant toute la durée du traitement et par la prise supplémentaire de mesna (uromitexan), qui se lie à l’acroléine, métabolite de la cyclophosphamide, et la neutralise.
Le rituximab peut entraîner une déplétion prolongée des lymphocytes B avec une immunodéficience humorale secondaire et des infections respiratoires sévères fréquentes, en particulier chez les patients ayant déjà reçu du cyclophosphamide [21].
Dans les formes particulièrement sévères et/ou réfractaires de l’AMP systémique, des mesures et des médicaments supplémentaires sont parfois utilisés, y compris la plasmaphérèse, l’administration d’immunoglobulines par voie intraveineuse, le mycophénolate mofétil (MMF), l’alemtuzumab, un anticorps anti-CD52 qui déploie les cellules B et T, la globuline antithymocytes, la 15-déoxyspergualine – un inhibiteur de la différenciation cellulaire – et la transplantation de cellules souches hématopoïétiques. Le succès de ces traitements intensifiés n’est pas bien établi et leur utilisation doit être réservée à des centres expérimentés.
Un nouveau mécanisme d’action très prometteur pour le traitement de l’AMP généralisée, ainsi que d’autres AAV, est l’inhibition du système du complément. L’Avacopan, un antagoniste oral du récepteur C5a, a montré des résultats très prometteurs dans les études de phase 3, ce qui rend très probable une approbation prochaine. Chez les patients atteints d’AMP généralisée et recevant de l’Avacopan en plus du traitement standard (cyclophosphamide ou rituximab), les glucocorticoïdes ont pu être arrêtés très rapidement. Il est intéressant de noter que la fonction rénale des patients traités par l’avacopan a montré une amélioration continue pendant toute la durée du traitement, soit 52 semaines [22].
Un médicament déjà disponible pour inhiber le système du complément est l’anticorps anti-C5 eculizumab (Soliris®), qui a été utilisé de manière isolée en cas d’évolution agressive d’une AAV [par ex. 24]. Une étude de phase 2 sur l’éculizumab dans la vascularite à ANCA a malheureusement été retirée avant l’inclusion des patients (NCT01275287). Un autre médicament, l’iptacopan (LNP023), qui inhibe l’activation efficace du système du complément via le facteur B, a récemment reçu l’autorisation de mise sur le marché pour le traitement de la glomérulopathie C3 (C3G), et il est fort possible que ce médicament soit également utilisé à l’avenir pour le traitement des AAV.
Maintien de la rémission en cas d’AMP généralisée. Après l’obtention d’une rémission clinique, le traitement de maintien de la rémission doit assurer l’activité inflammatoire de l’AAV même avec de petites doses de prednisone ou même sans glucocorticoïdes concomitants. Depuis 1995, l’European Vasculitis Study Group (EUVAS) a mené un grand nombre d’études d’intervention clinique sur l’AAV. Le MTX, l’azathioprine ou le rituximab sont recommandés pour le maintien de la rémission de l’AMP et des autres AAV. Le rituximab est peut-être le médicament le plus efficace des trois, bien que la dose optimale et l’intervalle entre les doses de rituximab dans l’AAV soient encore en discussion. Certains experts recommandent l’administration fixe de 1g de rituximab pendant 6 mois [24], tandis qu’une étude comparative française sur le rituximab contre l’azathioprine a montré de bons résultats pour un intervalle de 6 à 12 mois avec des doses de 500mg [25]. Sous rituximab, comme sous cyclophosphamide, les taux de PR3 ou de MPO-ANCA tombent en dessous du seuil de détection dans la plupart des cas, en corrélation avec la rémission clinique de la vascularite. Chez ces patients, notre expérience montre que l’intervalle de rituximab peut être adapté à la remontée des titres d’ANCA, ce qui permet dans certains cas d’étendre l’intervalle à plus de 12 mois.
Autres AAV (MPA, FNGN, EGPA)
MPA et FNGN
Contrairement à la GPA, la MPA et la FNGN ne présentent pas de granulomes. Ces AAV sont un peu plus souvent associés aux MPO-ANCA qu’aux PR3-ANCA, mais cela n’a pas d’importance pour le traitement. Les deux AAV se manifestent typiquement sous la forme d’une glomérulonéphrite “pauci-immune” à progression rapide (RPGN). Le terme “pauci-immun” désigne la faible, voire l’absence de détection des dépôts d’immunoglobulines lors de l’examen immunohistochimique des biopsies rénales. D’autres manifestations typiques de la MPA sont une mononévrite multiplex douloureuse et une alvéolite qui, chez environ un tiers des patients, peut conduire à une fibrose pulmonaire avec une mortalité nettement accrue [26]. Le traitement de la MPA et de la FNGN est similaire aux principes de traitement de la GPA généralisée décrits ci-dessus.
EGPA (anciennement : syndrome de Churg-Strauss/vascularite)
L’EGPA peut être considéré comme le “pendant atopique” de l’AMP. Elle se manifeste chez des patients présentant des symptômes atopiques, notamment de l’asthme et/ou une pansinusite chronique (figure 4) avec des polypes nasaux, depuis des années, voire des décennies. Pendant la phase initiale de l’EGPA, on trouve des granulomes dans les tissus concernés, comme dans le cas de l’AMP. Cependant, contrairement à la GPA, ces granulomes ne sont pas densément peuplés de granulocytes neutrophiles, mais de granulocytes éosinophiles. Par analogie, on trouve une éosinophilie dans le sang périphérique des patients EGPA. Sur le plan clinique, l’EGPA diffère de l’AMP sur des points importants. La survenue d’un RPGN est moins fréquente qu’avec la GPA (environ 15-20%) [27], mais comme pour la GPA, elle est strictement associée à la présence d’ANCA. Des manifestations neurologiques, notamment une mononévrite multiplex souvent très douloureuse [28], sont présentes chez plus de la moitié des patients. Les manifestations cardiaques de l’EGPA sont redoutables, par exemple la vascularite des artères coronaires, qui survient chez environ 40% des patients et constitue la principale cause de décès de l’EGPA [29,30].
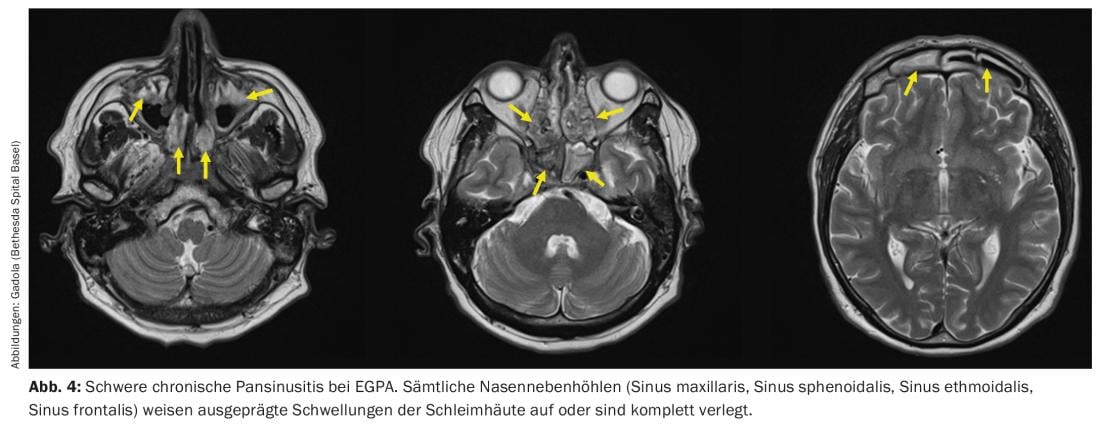
Le traitement de l’EGPA est très similaire à celui de l’AMP généralisée et les médicaments les plus fréquemment utilisés dans l’EGPA, comme dans l’AMP, sont les glucocorticoïdes, le MTX, le cyclophosphamide et le rituximab. Les glucocorticoïdes à forte dose entraînent très rapidement une baisse drastique des éosinophiles et une amélioration clinique. Cependant, comme pour l’AMP, la corticothérapie doit être rapidement réduite à la dose seuil de prednisone dans les 3 mois afin de réduire le risque d’infections graves des voies respiratoires. En cas d’EGPA récidivante ou résistante au traitement, on dispose en outre de l’interféron alpha (3×/semaine à administration quotidienne s.c.) et de l’anticorps anti-IL5 mépolizumab (Nucala®, 300 mg s.c. toutes les 4 semaines), qui est également autorisé pour cette indication. Si le traitement est efficace, l’éosinophilie devrait disparaître et le titre d’ANCA devrait diminuer.
Vascularites des petits vaisseaux non associées aux ANCA
Vascularite cryoglobulinémique essentielle
Cette vascularite des petits vaisseaux est causée par l’activation in situ de la cascade du complément dans les parois des vaisseaux après le dépôt de cryoglobulines de type II (plus rarement de type III). Elle se manifeste principalement au niveau de la peau par une vascularite urticarienne ou un purpura palpable (Fig. 5). En cas d’atteinte rénale, on observe généralement une glomérulonéphrite membranoproliférative à médiation par le complément et à complexe immunitaire de type lupique. D’autres symptômes typiques sont les myalgies et les arthralgies, ainsi qu’une polyneuropathie. Pendant les poussées, les compléments C4 (“toujours”) et C3 (“souvent”) sont abaissés dans le sérum.
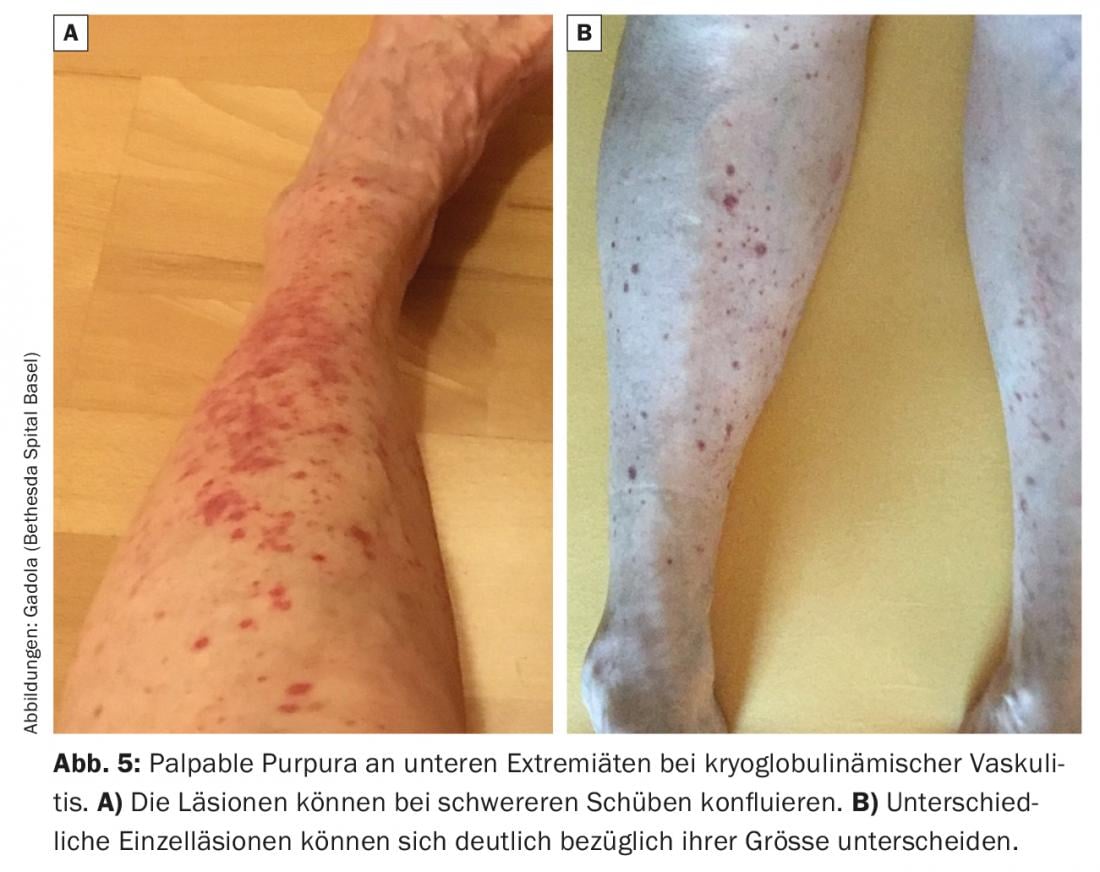
Le traitement commence par des glucocorticoïdes et est complété si nécessaire par des DMARD (MTX ou azathioprine). Dans les cas résistants au traitement, le rituximab s’est révélé très efficace, et dans les cas aigus graves avec une atteinte rénale sévère, le cyclophosphamide est utilisé [31].
Plus fréquentes que la vascularite cryoglobulinémique essentielle, dont l’étiologie est par définition inconnue, sont la vascularite cryoglobulinémique associée au VHC (cryoglobulines sériques de type II ou de type III) et les poussées de vascularite cryoglobulinémique liées à des néoplasies à cellules B (cryoglobulines de type I ou de type III). Dans ce cas, la priorité est donnée au traitement de la maladie sous-jacente et, dans les cas graves, au traitement de déplétion des cellules B par rituximab.
Vascularite leucocytoclasique
En général, la vascularite leucocytoclasique à médiation par complexe immunitaire est strictement limitée aux capillaires et aux veinules du derme et se manifeste par un purpura palpable prurigineux à douloureux des membres inférieurs. Si d’autres organes que la peau sont touchés, il faut toujours rechercher une autre vascularite des petits vaisseaux primaire ou secondaire. La vascularite leucocytoclasique limitée à la peau. est souvent autolimitée ; toutefois, des évolutions graves récurrentes ou chroniques peuvent survenir, qui sont traitées par des glucocorticoïdes et, le cas échéant, par différents DMARD ; dans les cas particulièrement graves, également par le cyclophosphamide.
Les autres vascularites “idiopathiques” (et donc “primaires”) des petits vaisseaux de l’adulte sont la vascularite urticarienne hypocomplémentémique associée aux anticorps anti-C1q (HUV), la vascularite à IgA (Henoch-Schönlein) et la vascularite anti-GBM (GBM, membrane basale glomérulaire). Le traitement de ces vascularites correspond en grande partie aux principes décrits ci-dessus pour les autres vascularites des petits vaisseaux.
Maladie de Behçet
La maladie de Behçet se caractérise par un large spectre clinique et une évolution par poussées. La vascularite de la maladie de Behçet peut toucher les vaisseaux artériels et/ou veineux de tous calibres. La vascularite des artères pulmonaires avec formation consécutive d’un anévrisme et rupture dans le tissu pulmonaire est la cause directe de décès la plus fréquente. Des complications graves sont en outre causées par des thromboses veineuses inflammatoires, une implication cardiaque et cérébrale.
La maladie de Behçet présente quelques similitudes cliniques frappantes avec la maladie de Crohn, comme l’apparition d’une colite, de fistules entérocolites et périanales, d’aphtes muco-cutanés, d’arthites, d’érythème noueux et d’uvéites. Le traitement des manifestations gastro-intestinales de la maladie de Behçet en est le reflet : outre les glucocorticoïdes, on utilise l’acide 5-aminosalycilique (5-ASA), l’azathioprine et, en cas de réponse insuffisante, des bloqueurs du TNFalpha. Pour le traitement des aphtes cutanéo-muqueux, on peut utiliser, selon la gravité, des stéroïdes topiques, la colchicine ou l’aprémilast (Otezla®), un inhibiteur de la phosphodiestérase-4 autorisé à cet effet. En cas d’atteinte oculaire aiguë, les glucocorticoïdes systémiques (par ex. 1 g de méthylprednisolone en IV en cas d’uvéite hypopionique) sont recommandés en première intention, toujours en association avec un DMARD, au premier rang desquels la ciclosporine A ou l’azathioprine, ou avec l’interféron-alpha ou un bloqueur du TNF alpha (infliximab ou adalimumab) [32]. En cas d’atteinte vasculaire des artères pulmonaires, les glucocorticoïdes à haute dose sont efficaces en association avec le cyclophosphamide ou les bloqueurs du TNF alpha. Les interventions non médicamenteuses sont également utilisées en cas de maladie de Behçet sévère. En cas de risque d’hémorragie d’un anévrisme de l’artère pulmonaire de grande taille, le traitement par embolisation est recommandé en premier lieu, plutôt que la révision vasculaire ouverte par chirurgie thoracique. En cas d’hémorragie gastro-intestinale grave, de menace de perforation intestinale ou de sténose intestinale, les patients doivent être opérés d’urgence.
Messages Take-Home
- Pour toutes les vascularites, le premier objectif du traitement est d’obtenir le plus rapidement possible une rémission clinique aussi complète que possible. Elle est suivie d’une phase de maintien de la rémission, qui peut durer plusieurs années.
- Le MTX ne doit jamais être associé au sulfométoxazole-trimétoprime dans le traitement du RZA, sous peine d’entraîner une myélosuppression sévère.
- L’inhibition du système du complément est un nouveau mécanisme d’action prometteur pour le traitement de l’AMP généralisée, ainsi que d’autres AAV.
- La vascularite systémique des petits vaisseaux dans l’AMP peut toucher tous les organes. L’atteinte des reins entraîne typiquement une glomérulonéphrite nécrosante “pauci-immune” (RPGN) à progression rapide, qui peut conduire à une insuffisance rénale grave si elle n’est pas traitée.
Littérature :
- Jardel S, et al : Mortalité dans les vascularites nécrotiques systémiques : une analyse rétrospective du registre du groupe d’étude français sur les vascularites. Autoimmun Rev 2018 Jul ; 17(7) : 653-659.
- Jennette JC, Falk RJ, Bacon PA, et al : 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013 Jan ; 65(1) : 1-11.
- Chung L, et al : Utilisation réussie du rituximab pour la vascularite cutanée. Arch Dermatol 2006 ; 142(11) : 1407-1410 ; doi:10.1001/archderm.142.11.1407.
- www.rheuma-net.ch/de/fachinformationen/behandlungsempfehlungen
- Villiger PM, Adler S, Kuchen S, et al : Tocilizumab for induction and maintenance of remission in giant cell arteritis : a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016 ; 387 : 1921-1927.
- Stone JH, Tuckwell K, Dimonaco S et al : Essai du tocilizumab dans l’artérite à cellules géantes. N Engl J Med 2017 ; 377 : 317-328.
- Christ L, et al : A Proof of Concept Study to Assess the Efficacy of Tocilizumab in Combination with Ultra-Short Glucocorticoid Administration to Treat Newly Diagnosed Giant Cell Arteritis – a 24 Week Analysis. Arthritis Rheumatol 2020 ; 72 (suppl 10).
- Adler S, et al : Risk of relapse after discontinuation of tocilizumab therapy in giant cell arteritis. Rheumatology (Oxford) 2019 Sep 1 ; 58(9) : 1639-1643.
- Hellmich B, et al : 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2020 Jan ; 79(1) : 19-30.
- Gadola SD, Gross WL : The renaissance of granulomatous inflammation in AAV. Nat Rev Rheumatol 2012 Jan 10 ; 8(2) : 74-76.
- Holle JU, et al : Les masses orbitales dans la granulomatose avec polyangéite sont associées à une évolution réfractaire et à un fardeau élevé de dommages locaux. Rhumatologie 2013 ; 52 : 875882.
- DeRemee RA, et al : Granulomatose de Wegener : observations sur le traitement par des agents antimicrobiens. Case Reports Mayo Clin Proc 1985 Jan ; 60(1) : 27-32 ; doi : 10.1016/s0025-6196(12)65279-3.
- Stegeman CA, et al : Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med 1994 Jan 1 ; 120(1) : 12-17.
- Rennie Rhee D, et al : Bactéries nasales associées à l’activité de la maladie et aux niveaux d’ANCA dans la granulomatose avec polyangéite. Arthritis Rheumatol 2020 ; 72 (suppl 10).
- Taylor GB, et al : A clinicopathological review of 34 cas of inflammatory breast disease showing an association between corynebacteria infection and granulomatous mastitis. Pathology 2003 Apr ; 35(2) : 109-119.
- Dobinson HC, et al : Options de traitement antimicrobien pour la mammite granulomateuse causée par des espèces de Corynebacterium. J Clin Microbiol 2015 Sep ; 53(9) : 2895-2899.
- Holle JU, Dubrau C, Herlyn K, et al : Rituximab pour la granulomatose réfractaire avec polyangéite (granulomatose de Wegener) : comparaison de l’efficacité dans les manifestations granulomateuses versus vasculaires. Ann Rheum Dis 2012 ; 71 : 327-333.
- Voswinkel J, Mueller A, Kraemer JA, et al : B lymphocyte maturation in Wegener’s granulomatosis : a comparative analysis of VH genes from endonasal lesions, Ann Rheum Dis 2006 ; 65 : 859-864.
- Voswinkel J, Assmann G, Held G et al : Single cell analysis of B lymphocytes from Wegener’s granulomatosis : B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol 2008 ; 154 : 339-345.
- Knight A, et al : Cancer urinaire de la vessie dans la granulomatose de Wegener : risques et relation avec le cyclophosphamide. Ann Rheum Dis 2004.
- Thiel J, et al : Cinétique de repopulation cellulaire B après traitement par rituximab dans les vascularites associées aux ANCA par rapport à l’arthrite rhumatoïde, et les maladies des tissus connectifs : une étude observationnelle longitudinale sur 120 patients. Arthritis Res Ther 2017 May 18 ; 19(1) : 101.
- Merkel P, et al : The Effect on Renal Function of the Complement C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. Arthritis Rheumatol 2020 ; 72 (suppl 10).
- Huizenga N, et al : Treatment of Aggressive Antineutrophil Cytoplasmic Antibody-Associated Vasculitis With Eculizumab. Kidney Int Rep 2020 Apr ; 5(4) : 542-545.
- Smtih R, et al : Extended Follow-Up of Patients Recruited to a Randomized, Controlled Trial of Rituximab versus Azathioprine After Induction of Remission with Rituximab for Patients with ANCA-Associated Vasculitis and Relapsing Disease. Arthritis Rheumatol 2020 ; 72 (suppl 10).
- Guillevin L, et al : Rituximab versus azathioprine pour l’entretien dans la vascularite associée à l’ANCA. N Engl J Med 2014 Nov 6 ; 371(19) : 1771-1780.
- Tzelepis GE, et al : Prévalence et issue de la fibrose pulmonaire dans la polyangéite microscopique. European Respiratory Journal 2010 ; 36 : 116-121.
- Sinico RA, et al : Implication rénale dans le syndrome de Churg-Strauss. Am J Kidney Dis 2006 May ; 47(5) : 770-779.
- Wolf J, et al. : Complications neurologiques d’un syndrome de Churg-Strauss : une étude prospective monocentrique. Aktuelle Neurologie 2009 ; 36 : V188.
- Conron M, Beynon HL : Syndrome de Churg-Strauss. Thorax 2000 Oct ; 55(10) : 870-877.
- Solans R, et al : Syndrome de Churg-Strauss : résultat et suivi à long terme de 32 patients. Rheumatology (Oxford) 2001 ; 40 : 763-771.
- Braun G, et al : Cryoglobulinaemic vasculitis : classification et aspects cliniques et thérapeutiques. Postgrad Med J 2007 Feb ; 83(976) : 87-94 ; doi : 10.1136/pgmj.2006.046078.
- Bettiol A, et al : Traiter les différents phénotypes du syndrome de Behçet. Front Immunol 2019 Dec 6 ; 10 : 2830 : doi : 10.3389/fimmu.2019.02830. eCollection 2019.
InFo DOULEUR & GERIATRIE 2020 ; 2(2) : 12-19









