
La perte de fonction du canal CFTR (Cystic fibrosis Transmembrane Conductance Regulator) entraîne la formation d’une sécrétion très visqueuse dans la mucoviscidose, ce qui entraîne une diminution de la clairance mucociliaire. Les patients signalent une toux chronique et des expectorations purulentes, ainsi qu’une augmentation des infections respiratoires. Le déplacement des sécrétions peut entraîner des atélectasies et d’autres complications telles que des pneumothorax ou des hémoptysies.
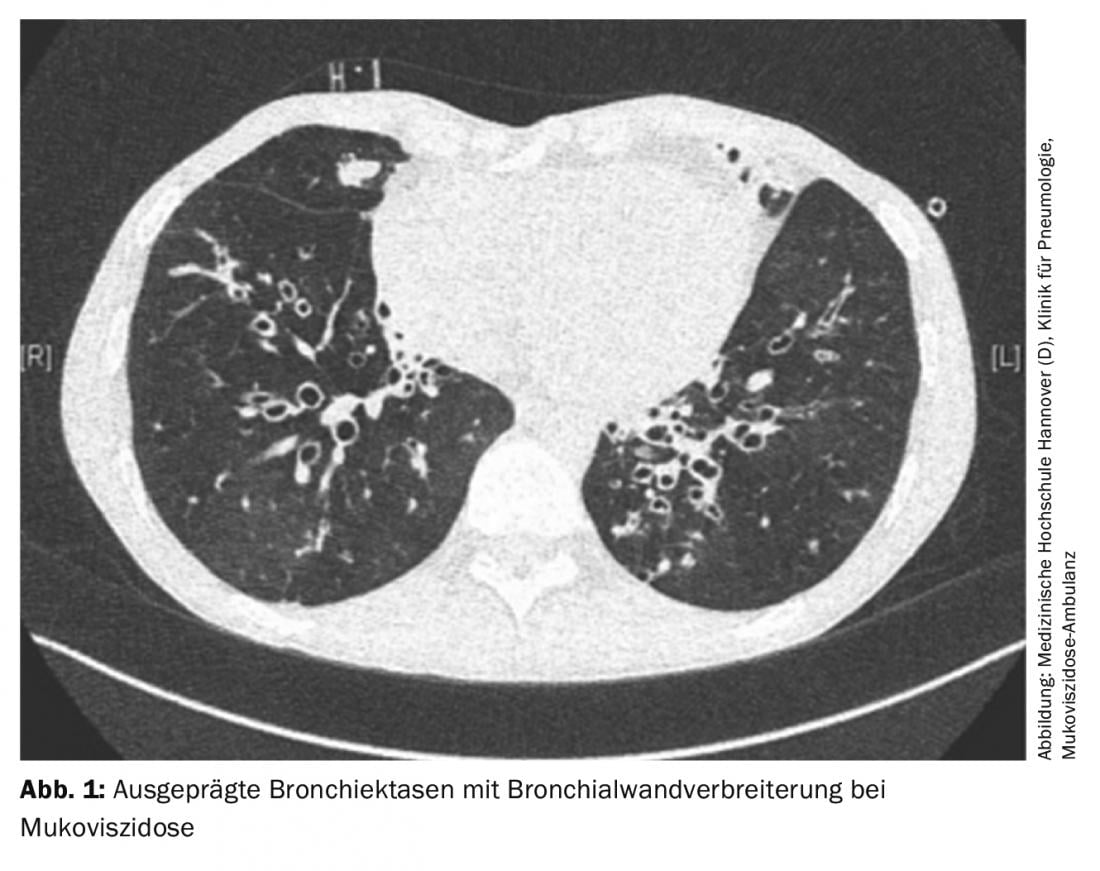
La perte de fonction du canal CFTR ( Cystic fibrosis Transmembrane Conductance Regul ator) entraîne la formation d’une sécrétion très visqueuse dans la mucoviscidose, ce qui entraîne une diminution de la clairance mucociliaire. Cela favorise la colonisation par des germes et entraîne une inflammation et une destruction du tissu pulmonaire, avec une insuffisance respiratoire consécutive. Sur le plan pulmonaire, la progression de la maladie se traduit généralement par un trouble ventilatoire obstructif avec hyperinflation. Les patients signalent une toux chronique et des expectorations purulentes, ainsi qu’une augmentation des infections respiratoires. L’imagerie montre souvent des bronchectasies remplies de mucus avec des parois bronchiques épaissies (figure 1). Les complications peuvent être des pneumothorax ou des hémoptysies.
En fonction de cette symptomatologie, le traitement de la maladie pulmonaire met l’accent sur la sécrétolyse, le traitement antibactérien et le traitement anti-inflammatoire.
Sécrétolyse
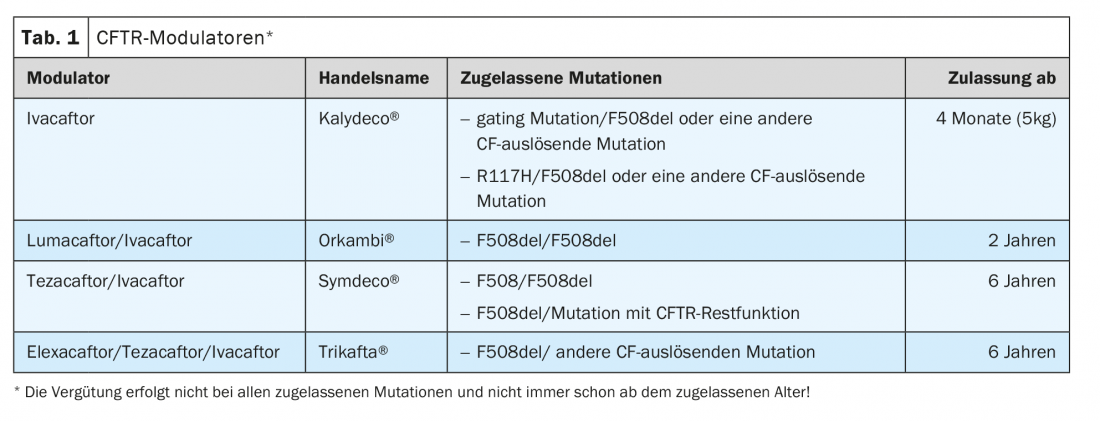
Modulateurs CFTR : le canal CFTR est un canal anionique pour le chlorure et le bicarbonate. Cette protéine membranaire est localisée dans l’épithélium respiratoire, mais aussi dans le tractus gastro-intestinal et dans de nombreux autres organes. Le premier modulateur de CFTR, l’ivacaftor, a été approuvé il y a 10 ans comme le premier traitement à agir sur le défaut de base sous-jacent de la mucoviscidose (tableau 1).
Ivacaftor
Parmi les modulateurs CFTR, on distingue les potentialisateurs et les correcteurs. Les potentialisateurs, dont l’ivacaftor fait partie, augmentent la probabilité d’ouverture du canal CFTR en activant des mécanismes indépendants de l’ATP. En conséquence, les études cliniques ont montré un effet sur les mutations pour lesquelles le canal ionique CFTR est présent dans la membrane cellulaire, mais dont la probabilité d’ouverture doit être augmentée. Le VEMS a augmenté de 10,6% points chez les patients présentant au moins une mutation G551D (c.1652G>A). La probabilité de développer une exacerbation pulmonaire a diminué de 55%, la qualité de vie et le poids ont augmenté.
Ces résultats ont conduit à l’approbation de l’ivacaftor en 2012 pour les mutations de Gating, qui sont toutefois rares, de sorte qu’environ 7% des patients atteints de mucoviscidose ont pu recevoir le médicament. Des études de registre menées au Royaume-Uni et aux États-Unis montrent que les effets positifs sont encore détectables 4 et 5 ans après le début du traitement.
Lumacaftor
Chez environ 50% des patients atteints de mucoviscidose, la mutation F508del est présente sous forme homozygote. Dans ce groupe très important, le traitement par ivacaftor s’était révélé inefficace. Des études de phase 2 ont montré qu’une combinaison d’ivacaftor et de lumacaftor pourrait entraîner une amélioration clinique. Le lumacaftor fait partie des correcteurs CFTR. En modifiant le processus dans la cellule, il augmente la quantité de protéine CFTR dans la membrane cellulaire.
Les études de phase 3 ont montré un effet significatif de cette thérapie combinée, même si cet effet était inférieur à celui de l’ivacaftor dans les mutations gating. Le VEMS a augmenté de 3 points et les exacerbations ont diminué de 30 %. Cette thérapie a été approuvée en 2016 pour ce groupe de patients.
Tezacaftor
Deux ans plus tard, les données relatives à un autre modulateur CFTR, le correcteur Tezacaftor combiné à l’ivacaftor, ont été publiées. Une étude de phase 3 contrôlée par placebo chez des patients présentant une mutation homozygote du F508del a montré une supériorité du traitement combiné de Tezacaftor et d’ivacaftor, avec une augmentation de 4% des points de VEMS. Là encore, le taux d’exacerbations pulmonaires était inférieur de 35% à celui du groupe placebo. L’effet était donc comparable à celui de l’orkambi chez les patients homozygotes F508del.
Une autre étude a comparé l’efficacité chez des patients présentant une F508del et une autre mutation CFTR avec fonction résiduelle. Ces patients ont été divisés en trois groupes : le premier groupe a reçu du Tezacaftor en combinaison avec de l’ivacaftor, le deuxième groupe de l’ivacaftor seul et le troisième groupe un placebo. Les deux groupes de traitement étaient supérieurs au groupe placebo en termes de VEMS. L’ivacaftor seul a entraîné une augmentation de 4,7 points, tandis que le traitement combiné a permis d’atteindre une augmentation de 6,8 points.
Sur la base de ces résultats, le traitement combiné a été approuvé à la fois pour les patients homozygotes F508del et pour les patients présentant une mutation F508del et une autre mutation CFTR avec fonction résiduelle.
Elexacaftor/Tezacaftor/Ivacaftor
Un autre grand pas en avant dans l’amélioration de la thérapie est démontré par les études cliniques sur l’efficacité de la triple combinaison d’Elexacaftor, Tezacaftor et Ivacaftor. Une étude contrôlée par placebo publiée en 2019 a été menée sur des patients présentant une mutation F508del et une autre mutation CFTR avec une fonction minimale (environ 200 mutations différentes).
Les résultats de cette étude ont largement dépassé les résultats précédents. La fonction pulmonaire a montré une augmentation du VEMS dès 2 semaines de traitement, qui est resté stable à 13,6% de points jusqu’à la semaine 24, et le taux d’exacerbations pulmonaires a été réduit de 60%. La qualité de vie des patients s’est nettement améliorée (18,1 points au CFQ-R), tout comme leur poids (IMC +1,3). Afin d’évaluer la fonction CFTR sous traitement, des tests de soudure ont été effectués. Il y a eu une baisse significative de 40 mmol/l de la concentration de chlorure de sueur, pathologiquement élevée dans la mucoviscidose, de sorte que de nombreux patients ne dépassaient plus le seuil de 60 mmol/l exigé pour le diagnostic de la mucoviscidose.
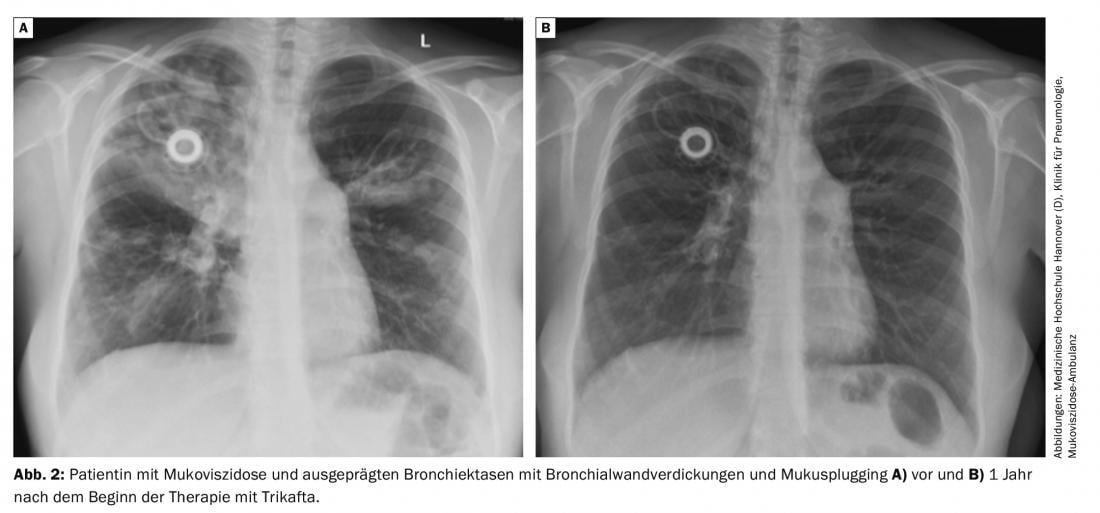
L’efficacité de la triple combinaison a également été testée chez des patients homozygotes F508del. Le traitement par Tezacaftor et Ivacaftor a servi de contrôle dans cette étude qui n’a duré que 4 semaines. La triple combinaison s’est avérée nettement supérieure au traitement par Tezacaftor et Ivacaftor, le VEMS a augmenté de 10 points, et des avantages significatifs ont également été observés en termes de qualité de vie et de teneur en chlorure du test de transpiration (Fig. 2).
Le traitement par Elexacaftor/Tezacaftor/Ivacaftor est généralement bien toléré. Les événements indésirables qui sont survenus plus fréquemment dans le cadre de l’étude clinique dans le groupe verum que dans le groupe placebo sont les éruptions cutanées (10%), l’augmentation du ferment hépatique (10%), les maux de tête et la diarrhée. 1% des patients ont interrompu le traitement en raison d’événements indésirables.
Les modulateurs CFTR doivent être pris avec un repas gras. Les patients ne doivent pas manger de pamplemousse ou d’orange amère ni prendre de millepertuis en raison d’interactions. La posologie doit être adaptée en cas d’utilisation simultanée d’inhibiteurs modérés ou puissants du CYP3A (par ex. antifongiques azolés, antidépresseurs).
Le coût d’un traitement par modulateurs CFTR se situe entre 12 000 et 17 000 CHF par mois. L’indication, l’initiation et le suivi du traitement doivent donc être effectués par un centre expérimenté dans le traitement de la fibrose kystique.
Derrière ces nombreux chiffres impressionnants se cache au quotidien un changement fondamental de la qualité et des perspectives de vie. De nombreux patients ne toussent plus et n’expectorent plus, des symptômes qui étaient jusqu’à présent des compagnons constants. Même s’il n’existe pas encore de données sur l’évolution à long terme, on peut supposer que ces médicaments augmentent considérablement l’espérance de vie.
Néanmoins, le médicament ne fonctionne pas de la même manière chez tous les patients et les patients dont les poumons sont gravement détruits continuent à avoir une fonction pulmonaire limitée. Le traitement n’est pas autorisé pour environ 10% des patients et environ 1 à 2% des patients arrêtent le traitement en raison d’intolérances. Par conséquent, d’autres formes de thérapie restent importantes.
Dornase alfa : dans le cadre de l’inflammation des voies respiratoires, l’ADN est libéré par la désintégration des granulocytes. La dornase alfa clive l’ADN et diminue ainsi la viscosité des sécrétions. Dans les études cliniques, cela a permis de réduire les exacerbations pulmonaires et d’améliorer la fonction pulmonaire.
Solution saline hypertonique et mannitol : par un effet osmotique, l’inhalation de solution saline hypertonique ou de mannitol entraîne une réduction de la viscosité des expectorations, ce qui améliore la clairance mucociliaire. Pour éviter une obstruction des voies respiratoires, un bronchodilatateur doit être utilisé avant l’inhalation.
Physiothérapie et sport : la physiothérapie et le sport font partie du traitement de base, même si les études cliniques prouvant leur efficacité sont insuffisantes. La thérapie permet d’apprendre des techniques pour améliorer la clairance mucociliaire, d’optimiser les techniques de toux et d’inhalation, de mobiliser le thorax et de faire des exercices de musculation et d’endurance.
Thérapies antibiotiques
Staphylocoque doré : chez les enfants et les adolescents, le staphylocoque doré est le germe le plus fréquemment détecté dans les sécrétions bronchiques. Il n’existe pas de stratégie unique pour traiter ce germe. Il existe des preuves que ce germe provoque une augmentation de l’inflammation des voies respiratoires et qu’il a donc une importance dans l’évolution de la maladie. Une tentative d’éradication par antibiotiques oraux est donc souvent effectuée, en particulier chez les enfants, même en cas de détection asymptomatique. En cas de détection symptomatique, un traitement antibiotique oral doit être administré dans tous les cas pendant 2 à 3 semaines.
Avec l’âge, on observe souvent une détection chronique du staphylocoque doré. Cependant, un traitement permanent à base de staphylocoques ne doit être envisagé que dans des cas exceptionnels, par exemple en cas d’exacerbations pulmonaires fréquentes, car il existe des preuves que ce traitement entraîne une augmentation de la détection de Pseudomonas.
Pseudomonas aeruginosa : avec l’âge, la fréquence de détection de Pseudomonas aeruginosa et d’autres germes à Gram négatif dans les sécrétions bronchiques augmente. Environ 30% des patients âgés de 18 ans et 75% des patients âgés de 45 à 49 ans sont colonisés de manière chronique par ce germe. Comme chaque nouvelle détection de germes ne s’accompagne pas d’une aggravation clinique, des frottis de gorge ou des échantillons d’expectoration doivent être examinés en routine environ 4x/an. La détection de Pseudomonas aeruginosa nécessite une longue période d’incubation, il faut donc s’assurer que le laboratoire de microbiologie remplit les conditions requises.
Dès la première détection de Pseudomonas aeruginosa, un traitement antibiotique par inhalation est indiqué pour l’éradication, indépendamment des symptômes cliniques. Différents protocoles équivalents sont disponibles à cet effet :
- Tobramycine 2× 300 mg pendant 4 semaines
- Colistine 2× 1 million d’UI pendant 3 mois en association avec ciprofloxacine 750 mg 2× 1 pendant 3 semaines
- Lysat d’aztréonam 3× 75 mg pendant 4 semaines.
L’éradication est considérée comme réussie si 6 contrôles ultérieurs sont négatifs. Cela réussit dans environ 80% des cas.
On parle de colonisation chronique par Pseudomonas aeruginosa lorsque sur 6 contrôles en 12 mois, plus de la moitié sont positifs. Dans ce cas, un traitement antibiotique permanent par inhalation est recommandé comme traitement de suppression. L’inhalation entraîne des niveaux d’expectoration plus élevés que l’administration i.v. et présente une toxicité moindre. Une bronchoconstriction après l’inhalation peut être contrecarrée par des bronchodilatateurs. Des inhalations de poudre sont également disponibles. Certes, l’inhalation provoque plus souvent de la toux que l’inhalation humide, mais le temps d’inhalation réduit constitue néanmoins un avantage évident pour de nombreux patients.
Les antibiotiques inhalés suivants sont disponibles :
Inhalation de poudre :
- Colistine 2× 1 gélule a 125 mg en continu
- Tobramycine 2× 4 gélules de 28 mg 28 jours on/off
Inhalation humide :
- Lysat d’aztréonam 3× 75 mg. 28 jours on/off
- Colistine 2× 1(-2) Mio UI en continu
- Levofloxacine 2× 240 mg 28 jours on/off (à partir de 18 ans)
- Tobramycine 2× 80/160/170/300 mg en continu ou 28 jours on/off
Exacerbation pulmonaire
Des exacerbations pumonales surviennent régulièrement chez les patients malgré le traitement suppressif. Elles se caractérisent généralement par plusieurs des symptômes suivants :
- Augmentation de la toux
- dyspnée croissante
- Modification de la quantité et de la couleur des crachats
- Fièvre
- sentiment de maladie, fatigue
- Perte de poids
- Chute de >10% du VEMS
- Augmentation des changements radiologiques
Une exacerbation pulmonaire peut être traitée par une intensification du traitement par inhalation, par un traitement antibiotique oral ou par un traitement antibiotique intraveineux.
Les médicaments oraux actifs contre les pseudomonas disponibles sont la ciprofloxacine et la lévofloxacine. Lors de la prescription, le risque de tendinite, voire de rupture du tendon, doit être pris en compte.
Un traitement intraveineux est généralement administré sous forme de thérapie combinée pendant 14 jours. Les médicaments les plus couramment utilisés sont la ceftazidime, le méropénème et la pipéracilline/tazobactam, associés à un aminoglycoside, généralement la tobramycine. Selon l’antibiogramme, d’autres antibiotiques peuvent être utilisés en cas d’augmentation de la résistance et d’absence d’effet des traitements standard.
Thérapie anti-inflammatoire
Les mesures de sécrétolyse et les traitements antibiotiques entraînent une réduction de l’inflammation. Le traitement par azithromycine constitue un autre traitement anti-inflammatoire. L’antibiotique macrolide, administré au long cours, entraîne une amélioration de la fonction pulmonaire et une réduction des exacerbations.
Un traitement à haute dose d’ibuprofène a montré des effets positifs dans les études cliniques, mais il ne s’est pas imposé dans la pratique clinique quotidienne en raison de la crainte d’effets secondaires gastro-intestinaux ou rénaux.
Les stéroïdes inhalés peuvent faire partie du traitement des patients souffrant d’asthme bronchique concomitant. Une utilisation générale en cas de mucoviscidose n’est pas indiquée. Il en va de même pour la stéroïdothérapie orale, qui n’est utilisée que chez les patients atteints d’aspergillose broncho-pulmonaire allergique (ABPA).
Transplantation pulmonaire
En cas de fonction pulmonaire sévèrement réduite, la transplantation pulmonaire représente une possibilité d’améliorer la qualité de vie et le pronostic à long terme. Il faut penser à se rendre au centre de transplantation dans les cas suivants :
- VEMS<30% ou en cas de chute rapide de la fonction pulmonaire
- exacerbations pulmonaires fréquentes, en particulier en cas de traitement en soins intensifs
- Pneumothoracées récurrentes
- hémoptysies répétées
Au niveau international, 15% des transplantations pulmonaires sont réalisées pour la mucoviscidose. Il est probable que ce nombre diminue considérablement grâce à l’amélioration des options thérapeutiques offertes par les modulateurs de la CFTR.
Polypose nasale et sinusite chronique
De nombreux patients atteints de mucoviscidose souffrent de sinusite chronique et de polypose nasale. La souffrance causée par l’obstruction nasale, la douleur au-dessus des NNH et la diminution de l’odorat est souvent très importante.
Comme traitement local conservateur, la douche nasale avec du NaCl 0,9% est la première mesure à prendre pour éliminer les sécrétions et les croûtes. Il est également possible d’utiliser des inhalateurs, dont certains produisent en outre une vibration afin d’obtenir une meilleure déposition dans les sinus. Les stéroïdes topiques sont particulièrement efficaces pour les polypes nasaux associés à des allergies, un essai thérapeutique est approprié chez les patients atteints de mucoviscidose, même si l’inflammation des neutrophiles est généralement au premier plan. Les α-sympathomimétiques topiques ne doivent être utilisés que rarement en raison de la tachyphylaxie.
L’effet des modulateurs de la CFTR sur la polypose nasale n’a pas été évalué dans les études d’enregistrement. Des études publiées entre-temps montrent qu’il y a également une nette diminution des plaintes dans ce domaine.
Si le traitement conservateur ne permet pas d’améliorer les symptômes de manière satisfaisante, un traitement chirurgical est possible. Cependant, les récidives sont très fréquentes après l’ablation des polypes seuls.
Insuffisance pancréatique exocrine, pancréatites récidivantes
Environ 85% des patients atteints de mucoviscidose souffrent d’une insuffisance pancréatique exocrine. Les douleurs abdominales qui en résultent, les selles massives et malodorantes avec des dépôts de graisse et la malnutrition constituent un symptôme majeur précoce de la maladie. L’insuffisance pancréatique exocrine existe généralement dès la naissance. Le traitement consiste en une substitution des enzymes pancréatiques au moment du repas.
Les pancréatites récidivantes surviennent chez environ 20% des patients qui ne présentent pas d’insuffisance pancréatique exocrine. Il s’agit souvent de patients dont l’évolution est plutôt bénigne en termes de maladie pulmonaire.
Le traitement des pancréatites est symptomatique. Des rapports de cas isolés suggèrent que les modulateurs de CFTR peuvent avoir une influence positive sur l’apparition de pancréatites. D’autre part, la survenue de pancréatites après le début d’un traitement par modulateur de la CFTR a été décrite.
Complications hépatobiliaires
Environ 40% des patients développent une fibrose biliaire focale, qui évolue vers une cirrhose chez environ 20% de ces patients. Les options thérapeutiques sont très limitées. L’acide ursodésoxycholique est recommandé dans les lignes directrices européennes, mais les données à ce sujet sont faibles. Dans certains cas, une transplantation hépatique peut être nécessaire. On ne sait pas encore comment les modulateurs de CFTR influencent le développement de la fibrose ou de la cirrhose.
Malignomes
Le risque de tumeurs gastro-intestinales est plus élevé chez les patients atteints de mucoviscidose. La réalisation d’une coloscopie en tant qu’examen de dépistage est donc recommandée à partir de l’âge de 40 ans.
Diabète sucré
La prévalence du diabète sucré dans la mucoviscidose augmente considérablement avec l’âge et atteint environ 30% à l’âge de 30 ans.
Le traitement se fait généralement par insuline. Une étude clinique a montré qu’au moins pendant les deux premières années suivant le diagnostic, un traitement oral par répaglinide n’est pas inférieur à une insulinothérapie. Il n’est pas possible d’évaluer de manière définitive dans quelle mesure le traitement modulateur de la CFTR influence l’apparition d’un diabète sucré. Là aussi, il existe des rapports individuels positifs.
Infertilité masculine
98% des hommes atteints de mucoviscidose sont infertiles en raison d’une aplasie bilatérale congénitale des vaisseaux déférents (CBAVD), qui s’est déjà produite en prénatal et qui entraîne une azoospermie. Comme la production de spermatozoïdes est généralement préservée, les spermatozoïdes peuvent être prélevés dans les testicules ou l’épididyme et utilisés pour une insémination intra-utérine (IIU), une fécondation in vitro (FIV) ou une injection intracytoplasmique de spermatozoïdes (ICSI).
Aspects gynécologiques chez les patientes atteintes de mucoviscidose
Chez les patientes atteintes de mucoviscidose, il n’y a qu’une légère diminution de la fertilité, a.e. due à l’augmentation de la viscosité des sécrétions cervicales. De nombreux rapports de cas suggèrent que la normalisation de la viscosité par le traitement avec des modulateurs CFTR entraîne une amélioration de la fertilité.
La question de savoir si le traitement par un modulateur de la CFTR doit être poursuivi en cas de grossesse doit être examinée au cas par cas. Les modulateurs de la CFTR traversent la barrière placentaire, mais aucun effet négatif sur l’enfant n’a été décrit. Les études sur les animaux n’ont pas non plus révélé d’effets nocifs.
Littérature :
- Ramsey BW, et al : A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011 ; 365(18) : 1663-1672.
- Volkova N, et al : Progression de la maladie chez les patients atteints de mucoviscidose traités par ivacaftor : données issues des registres nationaux américain et britannique. JCF 2020 ; 10 : 68-70.
- Wainwright CE, et al : Tezacaftor-Ivacaftor chez les patients atteints de mucoviscidose homozygote pour Phe508del CFTR. N Engl J Med 2015 ; 373(3) : 220-231.
- Taylor-Cousar JL, et al : Tezacaftor-Ivacaftor chez les patients atteints de mucoviscidose homozygote pour Phe508del. N Engl J Med 2017 ; 377(21) : 2013-2023.
- Rowe SM, et al : Tezacaftor-Ivacaftor dans les hétérozygotes à fonction résiduelle atteints de mucoviscidose. N Engl J Med 2017 ; 377(21) : 2024-2035.
- Middleton et al. Elexacaftor-Tezacaftor-Ivacftor pour la mucoviscidose avec un seul allèle Phe508del. N Engl J Med 2019 ; 381(19) : 1809-1819.
- Yang C, Montgomery M. : Dornase alfo for Cystic fibrosis. Cochrane Database Syst Rev 2021 ; 3(3) : CD001127 ; doi : 10.1002/14651858.CD001127.pub5.
- Wark P, McDonald VM. : Nebulized hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev 2018 ; 9(9) : CD001506 ; doi : 10.1002/14651858.CD001506.pub4.
- Beswick DM, et al : Impact de la thérapie du régulateur de conductance transmembranaire de la mucoviscidose sur la rhinosinusite chronique et l’état de santé : Deep Learning CT Analysis and Patient-reported Outcomes. Ann Am Thorac Soc 2022 ; 19(1) : 12-19.
- Hewer SCL, et al : Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis : Cochrane Database Syst Rev 2017 ; 4(4) : CD004197.
InFo PNEUMOLOGIE & ALLERGOLOGIE 2022 ; 4(4) : 14-18









