
La perdita di funzione del canale del regolatore di conduttanza transmembrana della fibrosi cistica (CFTR) nella fibrosi cistica comporta la formazione di una secrezione altamente viscosa che porta a una riduzione della clearance mucociliare. I pazienti riferiscono tosse cronica ed espettorato purulento, nonché un aumento delle infezioni respiratorie. L’atelettasia può verificarsi a causa dell’ostruzione delle secrezioni; ulteriori complicazioni possono includere pneumotoraci o emottisi.
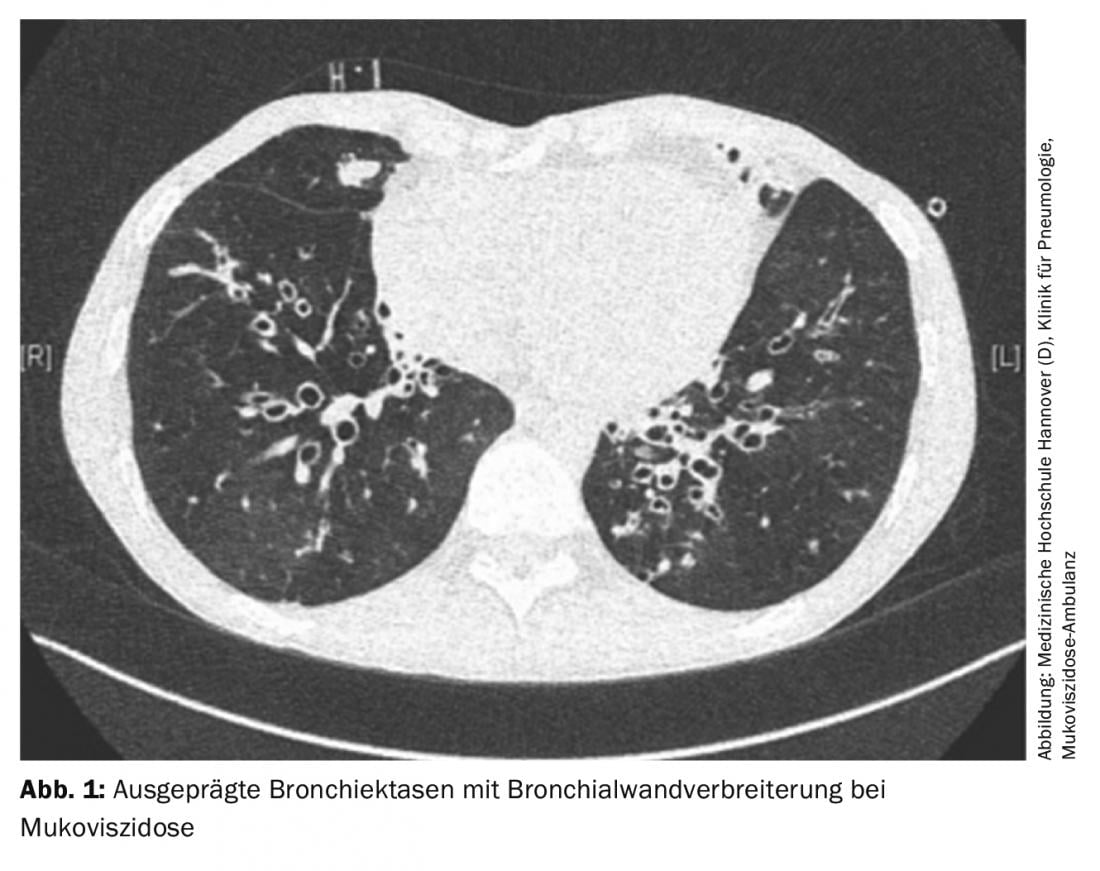
La perdita di funzione del canale del regolatore di conduttanza transmembrana della fibrosi cistica (CFTR) nella fibrosi cistica comporta la formazione di una secrezione altamente viscosa che porta a una riduzione della clearance mucociliare. Questo favorisce la colonizzazione batterica, portando all’infiammazione e alla distruzione del tessuto polmonare con conseguente insufficienza respiratoria. Dal punto di vista funzionale, con il progredire della malattia, di solito si verifica un disturbo ostruttivo della ventilazione con iperinflazione. I pazienti riferiscono tosse cronica ed espettorato purulento, nonché un aumento delle infezioni respiratorie. La diagnostica per immagini mostra spesso bronchiectasie piene di muco con pareti bronchiali ispessite (Fig. 1). L’atelettasia può verificarsi a causa dell’ostruzione delle secrezioni; ulteriori complicazioni possono includere pneumotoraci o emottisi.
In base a questi sintomi, il trattamento della malattia polmonare si concentra sulla secretolisi, sulla terapia antibatterica e antinfiammatoria.
Secretolisi
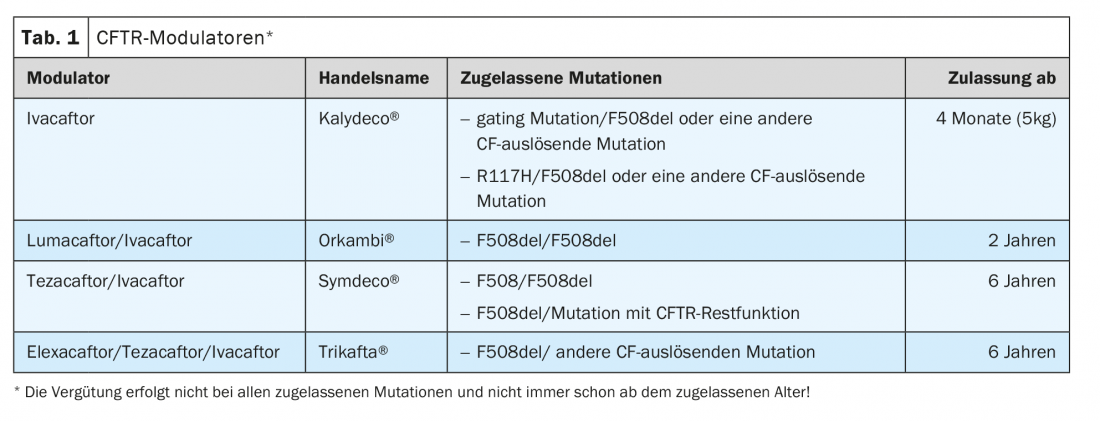
Modulatori CFTR: il canale CFTR è un canale anionico per il cloruro e il bicarbonato. Questa proteina legata alla membrana è localizzata nell’epitelio respiratorio, ma anche nel tratto gastrointestinale e in molti altri organi. Il primo modulatore CFTR, ivacaftor, è stato approvato 10 anni fa come prima terapia che agisce sul difetto di base della fibrosi cistica (Tabella 1).
Ivacaftor
I modulatori CFTR si distinguono tra potenziatori e correttori. I potenziatori, tra cui ivacaftor, aumentano la probabilità di apertura del canale CFTR attivando meccanismi indipendenti dall’ATP. Di conseguenza, gli studi clinici hanno mostrato un effetto nelle mutazioni in cui il canale ionico CFTR è presente nella membrana cellulare, ma la probabilità di apertura deve essere aumentata. Il FEV1 è aumentato di 10,6% punti nei pazienti con almeno una mutazione G551D (c.1652G>A). La probabilità di avere un’esacerbazione polmonare è diminuita del 55%, e la qualità della vita e il peso sono aumentati.
Questi risultati hanno portato all’approvazione dell’ivacaftor nel 2012 per le mutazioni di gating, che però sono rare, per cui circa il 7% dei pazienti con FC potrebbe ricevere il farmaco. Gli studi di registro della Gran Bretagna e degli Stati Uniti mostrano che gli effetti positivi sono ancora rilevabili rispettivamente 4 e 5 anni dopo l’inizio della terapia.
Lumacaftor
In circa il 50% dei pazienti con FC, la mutazione F508del è omozigote. In questo gruppo molto numeroso, la terapia con ivacaftor non si è dimostrata efficace. Gli studi di fase 2 hanno indicato che la combinazione di ivacaftor con lumacaftor può produrre un miglioramento clinico. Lumacaftor appartiene ai correttori CFTR. Aumenta la quantità di proteina CFTR nella membrana cellulare, modificando l’elaborazione nella cellula.
Negli studi di fase 3, questa terapia combinata ha mostrato un effetto significativo, anche se minore rispetto all’effetto di ivacaftor sulle mutazioni di gating. Il FEV1 è aumentato del 3% e le esacerbazioni sono diminuite del 30%. Questa terapia è stata approvata per questo gruppo di pazienti nel 2016.
Tezacaftor
Due anni dopo, sono stati pubblicati i dati su un altro modulatore CFTR, il correttore tezacaftor in combinazione con ivacaftor. In uno studio di fase 3 controllato con placebo su pazienti con mutazione F508del omozigote, il trattamento combinato di tezacaftor e ivacaftor ha dimostrato la superiorità con un aumento del 4% del FEV1. Anche in questo caso, il tasso di esacerbazioni polmonari è stato inferiore del 35% rispetto al gruppo placebo. L’effetto era quindi paragonabile a quello di Orkambi nei pazienti omozigoti F508del.
Un altro studio ha confrontato l’efficacia nei pazienti con F508del e un’altra mutazione CFTR con funzione residua. Questi pazienti sono stati divisi in tre gruppi: un gruppo ha ricevuto tezacaftor in combinazione con ivacaftor, il secondo gruppo ha ricevuto solo ivacaftor e il terzo gruppo ha ricevuto un placebo. Entrambi i gruppi di trattamento erano superiori al gruppo placebo in termini di FEV1. L’Ivacaftor da solo ha portato a un aumento di 4,7% punti, mentre il trattamento combinato ha ottenuto un aumento di 6,8% punti.
Sulla base di questi risultati, il trattamento combinato è stato approvato sia per i pazienti omozigoti F508del che per i pazienti che presentano F508del e un’altra mutazione CFTR con funzione residua.
Elexacaftor/Tezacaftor/Ivacaftor
Un altro grande passo avanti nel miglioramento della terapia è evidente negli studi clinici sull’efficacia della tripla combinazione di elexacaftor, tezacaftor e ivacaftor. In uno studio controllato con placebo pubblicato nel 2019, sono stati studiati i pazienti che presentavano una F508del e un’altra mutazione CFTR con funzione minima (circa 200 mutazioni diverse).
I risultati di questo studio hanno chiaramente superato le scoperte precedenti. La funzione polmonare ha mostrato un aumento del FEV1 dopo solo 2 settimane di terapia, che è rimasto stabile a 13,6% punti fino alla settimana 24, e il tasso di esacerbazioni polmonari è stato ridotto del 60%. La qualità di vita dei pazienti è aumentata in modo significativo (18,1 punti nel CFQ-R), così come il loro peso (BMI +1,3). Per valutare la funzione CFTR durante la terapia, sono stati eseguiti dei test del sudore. C’è stato un calo significativo di 40 mmol/l nella concentrazione di cloruro di sudore, che è patologicamente aumentata nella fibrosi cistica, cosicché molti pazienti non erano più al di sopra del limite di 60 mmol/l, richiesto per la diagnosi di fibrosi cistica.
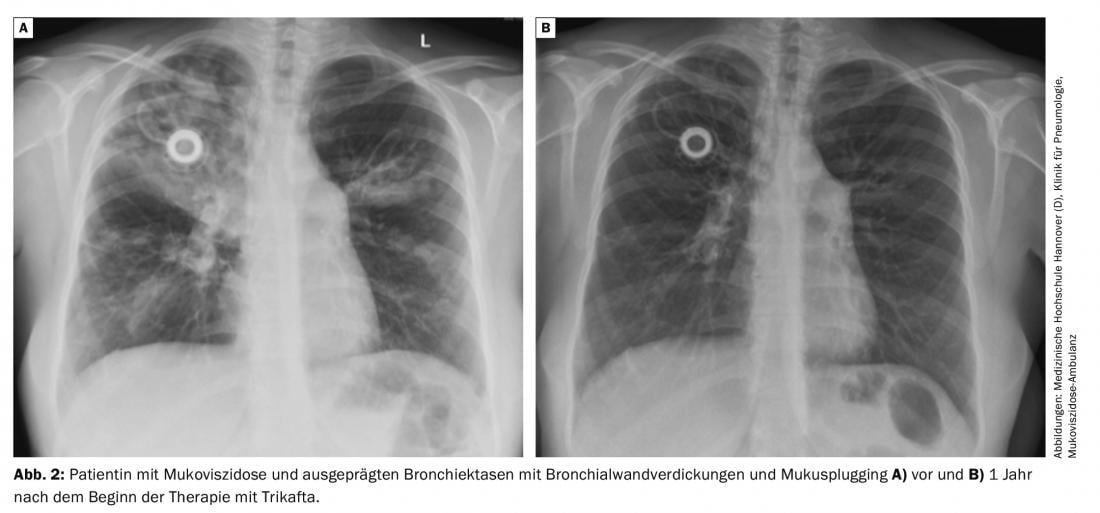
L’efficacia della tripla combinazione è stata testata anche nei pazienti omozigoti F508del. La terapia con Tezacaftor e ivacaftor è servita come controllo in questo studio, che è durato solo 4 settimane. La tripla combinazione è stata chiaramente superiore alla terapia con tezacaftor e ivacaftor, il FEV1 è aumentato del 10% di punti, e ci sono stati anche chiari vantaggi nella qualità della vita e nel contenuto di cloruro del test del sudore (fig. 2) .
La terapia con elexacaftor/tezacaftor/ivacaftor è solitamente ben tollerata. Gli eventi avversi che si sono verificati più frequentemente nel gruppo verum rispetto al gruppo placebo durante lo studio clinico sono stati eruzioni cutanee (10%), aumento degli enzimi epatici (10%), mal di testa e diarrea. L’1% dei pazienti ha interrotto la terapia a causa di eventi avversi.
I modulatori CFTR devono essere assunti con un pasto contenente grassi. I pazienti non devono mangiare pompelmo o arance amare o assumere l’erba di San Giovanni a causa delle interazioni, e il dosaggio deve essere adattato in caso di uso contemporaneo di inibitori moderati o forti del CYP3A (ad esempio, antifungini azolici, antidepressivi).
Il costo della terapia con i modulatori CFTR è di 12.000-17.000 franchi svizzeri al mese. L’indicazione, l’avvio e il monitoraggio della terapia devono quindi essere effettuati da un centro con esperienza nel trattamento della fibrosi cistica.
Dietro a queste cifre impressionanti si nasconde un cambiamento fondamentale nella qualità della vita e nella prospettiva di vita nella quotidianità. Molti pazienti non hanno più tosse o espettorato, sintomi che prima erano compagni costanti. Anche se non ci sono dati sul decorso a lungo termine della malattia, si può ipotizzare che l’aspettativa di vita sia significativamente aumentata da questi farmaci.
Tuttavia, il farmaco non funziona allo stesso modo in tutti i pazienti e i pazienti con polmoni gravemente distrutti continuano ad avere una funzione polmonare compromessa. La terapia non è approvata per circa il 10% dei pazienti e circa l’1-2% dei pazienti interrompe la terapia a causa di intolleranze. Pertanto, altre forme di terapia sono ancora importanti.
Dornase alfa: nel contesto dell’infiammazione delle vie respiratorie, il DNA viene rilasciato attraverso la disintegrazione dei granulociti. La dornasi alfa taglia il DNA e riduce così la viscosità della secrezione. Negli studi clinici, ciò ha comportato una riduzione delle esacerbazioni polmonari e un miglioramento della funzione polmonare.
Salina ipertonica e mannitolo: un effetto osmotico porta a una riduzione della viscosità dell’espettorato dopo l’inalazione di salina ipertonica o mannitolo, con conseguente miglioramento della clearance mucociliare. Per evitare l’ostruzione delle vie aeree, è necessario utilizzare un broncodilatatore prima dell’inalazione.
Fisioterapia e sport: la fisioterapia e lo sport fanno parte della terapia di base, anche se non ci sono studi clinici sufficienti a dimostrare la loro efficacia. La terapia prevede l’apprendimento di tecniche per migliorare la clearance mucociliare, l’ottimizzazione della tosse e della tecnica di inalazione, la mobilizzazione del torace e l’allenamento della forza e della resistenza.
Terapie antibiotiche
Staphylococcus aureus: nei bambini e negli adolescenti, lo Staphylococcus aureus è il germe più frequentemente rilevato nelle secrezioni bronchiali. Non esiste una strategia uniforme per il trattamento di questo germe. Ci sono indicazioni che questo germe provochi un’infiammazione maggiore delle vie respiratorie e che quindi abbia un’importanza per il decorso della malattia. Un tentativo di eradicazione con antibiotici orali viene quindi spesso effettuato, soprattutto nei bambini, anche in caso di rilevamento asintomatico. In caso di rilevamento sintomatico, si deve comunque somministrare una terapia antibiotica orale per 2-3 settimane.
Con l’avanzare dell’età, si osserva spesso un’evidenza cronica di Staphylococcus aureus. Tuttavia, la terapia stafilococcica a lungo termine deve essere presa in considerazione solo in casi eccezionali, ad esempio in caso di frequenti esacerbazioni polmonari, in quanto vi sono indicazioni che questa terapia porti ad una maggiore evidenza di Pseudomonas.
Pseudomonas aeruginosa: con l’aumentare dell’età, aumenta la frequenza di rilevamento di Pseudomonas aeruginosa e altri germi gram-negativi nelle secrezioni bronchiali. Circa il 30% dei pazienti di 18 anni e il 75% di quelli di 45-49 anni sono cronicamente colonizzati da questo germe. Poiché non tutti i nuovi germi rilevati sono accompagnati da un peggioramento clinico, i tamponi della gola o i campioni di espettorato devono essere esaminati di routine circa 4 volte all’anno. Il rilevamento di Pseudomonas aeruginosa richiede un lungo periodo di incubazione, pertanto bisogna assicurarsi che il laboratorio microbiologico soddisfi i requisiti adeguati.
Al primo rilevamento di Pseudomonas aeruginosa, è indicata una terapia antibiotica inalatoria per l’eradicazione, indipendentemente dai sintomi clinici. A questo scopo, sono disponibili diversi protocolli equivalenti:
- Tobramicina 2× 300 mg per 4 settimane
- Colistina 2× 1 milione di UI per 3 mesi in combinazione con ciprofloxacina 750 mg 2× 1 per 3 settimane.
- Lisato di aztreonam 3× 75 mg per 4 settimane.
L’eradicazione è considerata riuscita se 6 controlli successivi sono negativi. Questo riesce in circa l’80% dei casi.
Si parla di colonizzazione cronica da Pseudomonas aeruginosa se più della metà dei 6 controlli in 12 mesi sono positivi. In questo caso, si raccomanda una terapia inalatoria antibiotica permanente come trattamento di soppressione. L’inalazione determina livelli di espettorato più elevati rispetto alla somministrazione i.v. e presenta una tossicità inferiore. La broncocostrizione dopo l’inalazione può essere contrastata dai broncodilatatori. Sono disponibili anche inalazioni in polvere. Sebbene l’inalazione porti alla tosse più spesso rispetto all’inalazione umida, il tempo di inalazione ridotto è un chiaro vantaggio per molti pazienti.
Sono disponibili i seguenti antibiotici per via inalatoria:
Inalazione di polvere:
- Colistina 2× 1 capsula rigida da 125 mg continua
- Tobramicina 2× 4 capsule rigide à 28 mg 28 giorni on/off
Inalazione umida:
- Lisato di aztreonam 3× 75 mg. 28 giorni on/off
- Colistina 2× 1(-2) milioni di UI continua
- Levofloxacina 2× 240 mg 28 giorni on/off (a partire dai 18 anni)
- Tobramicina 2× 80/160/170/300 mg ininterrottamente o 28 giorni on/off
Esacerbazione polmonare
Più volte, i pazienti sperimentano esacerbazioni pumonali nonostante il trattamento di soppressione. Di solito sono caratterizzati da diversi dei seguenti sintomi:
- Aumento della tosse
- aumento della dispnea
- Cambiamento del volume e del colore dell’espettorato
- Febbre
- Sensazione di malessere, stanchezza
- Perdita di peso
- Diminuzione del FEV1 di >10%
- Aumento delle alterazioni radiologiche
L’esacerbazione polmonare può essere trattata intensificando la terapia inalatoria, con una terapia antibiotica orale o con una terapia antibiotica endovenosa.
La ciprofloxacina e la levofloxacina sono disponibili come farmaci orali, attivi contro lo pseudomonas. Al momento della prescrizione, bisogna considerare il rischio di tendinite o addirittura di rottura del tendine.
La terapia endovenosa viene solitamente somministrata come terapia combinata nell’arco di 14 giorni. I farmaci più comunemente utilizzati sono la ceftazidima, il meropenem e la piperacillina/tazobactam, che vengono combinati con un aminoglicoside – solitamente la tobramicina. A seconda dell’antibiogramma, si possono utilizzare altri antibiotici in caso di aumento della resistenza e di mancanza di effetto delle terapie standard.
Terapia antinfiammatoria
Le misure per la secretolisi e le terapie antibiotiche portano a una riduzione dell’infiammazione. Un’altra terapia antinfiammatoria è il trattamento con azitromicina. L’antibiotico macrolide porta a un miglioramento della funzione polmonare e a una riduzione delle esacerbazioni come terapia a lungo termine.
La terapia ad alto dosaggio con ibuprofene ha mostrato effetti positivi negli studi clinici, ma non si è affermata nella pratica clinica a causa delle preoccupazioni sugli effetti collaterali gastrointestinali o renali.
Gli steroidi per via inalatoria possono far parte della terapia nei pazienti con asma bronchiale coesistente. L’uso generale nella fibrosi cistica non è indicato. Lo stesso vale per la terapia steroidea orale, che viene utilizzata solo nei pazienti con aspergillosi broncopolmonare allergica (ABPA).
Trapianto di polmone
Nei casi di funzionalità polmonare gravemente compromessa, il trapianto di polmone è un modo per migliorare la qualità della vita e la prognosi a lungo termine. Una presentazione al centro trapianti dovrebbe essere presa in considerazione nei seguenti casi:
- FEV1<30% o in caso di rapido declino della funzione polmonare
- frequenti esacerbazioni polmonari, soprattutto. per il trattamento di terapia intensiva
- Pneumotoraci ricorrenti
- emottisi ripetuta
A livello internazionale, il 15% dei trapianti di polmone viene eseguito per la fibrosi cistica. Si può ipotizzare che questo numero diminuirà in modo significativo grazie alle migliori opzioni terapeutiche fornite dai modulatori CFTR.
Poliposi nasi e sinusite cronica
Molti pazienti affetti da fibrosi cistica soffrono di sinusite cronica e poliposi nasi. Il disagio causato dall’ostruzione nasale, dal dolore sopra le fosse nasali e dalla ridotta capacità di sentire gli odori è spesso molto significativo.
Come terapia conservativa e locale, l’irrigazione nasale con NaCl 0,9% per rimuovere le secrezioni e le croste è al primo posto. Possono essere utilizzati anche dispositivi di inalazione, alcuni dei quali generano anche una vibrazione per ottenere una migliore deposizione nei seni paranasali. Gli steroidi topici sono particolarmente efficaci per i polipi nasali associati alle allergie e una prova di terapia è appropriata nei pazienti con fibrosi cistica, anche se l’infiammazione neutrofila è di solito la preoccupazione principale. Gli α-simpaticomimetici topici devono essere usati raramente a causa della tachifilassi.
L’effetto dei modulatori CFTR sulla poliposi nasi non è stato indagato negli studi pivotal. Gli studi pubblicati nel frattempo dimostrano che c’è anche una riduzione significativa dei reclami in questo settore.
Se i sintomi non migliorano in modo soddisfacente con la terapia conservativa, è possibile ricorrere al trattamento chirurgico. Tuttavia, le recidive si verificano molto spesso dopo la sola rimozione dei polipi.
Insufficienza pancreatica esocrina, pancreatite ricorrente
Circa l’85% dei pazienti con FC presenta un’insufficienza pancreatica esocrina. Questo provoca dolori addominali, feci voluminose e maleodoranti con depositi di grasso e malnutrizione, che sono i primi sintomi della malattia. L’insufficienza pancreatica esocrina è solitamente presente alla nascita. La terapia consiste nella sostituzione degli enzimi pancreatici durante i pasti.
La pancreatite ricorrente si verifica in circa il 20% dei pazienti che non hanno un’insufficienza pancreatica esocrina. Spesso si tratta di pazienti che hanno un decorso piuttosto lieve in termini di malattia polmonare.
Il trattamento della pancreatite è sintomatico. Singoli rapporti di casi suggeriscono che i modulatori CFTR possono avere un’influenza positiva sull’insorgenza della pancreatite. D’altra parte, è stata descritta la comparsa di pancreatite dopo l’inizio della terapia con modulatore CFTR.
Complicazioni epatobiliari
In circa il 40% dei pazienti, si sviluppa una fibrosi biliare focale, che progredisce fino alla cirrosi in circa il 20% di questi pazienti. Le opzioni terapeutiche sono molto limitate. L’acido ursodesossicolico è raccomandato nelle linee guida europee, ma i dati sono deboli. In alcuni casi, può essere necessario un trapianto di fegato. Come i modulatori CFTR influenzino lo sviluppo della fibrosi o della cirrosi non è ancora noto.
Tumori maligni
I pazienti con fibrosi cistica hanno un rischio maggiore di tumori gastrointestinali. L’esecuzione di una colonscopia come esame preventivo è quindi raccomandata a partire dai 40 anni.
Diabete mellito
La prevalenza del diabete mellito nella fibrosi cistica aumenta significativamente con l’età e si aggira intorno al 30% all’età di 30 anni.
Il trattamento è di solito con l’insulina. Uno studio clinico ha dimostrato che, almeno nei primi due anni dopo la diagnosi, la terapia orale con repaglinide non è inferiore alla terapia insulinica. La misura in cui la terapia con modulatore CFTR influisce sullo sviluppo del diabete mellito non può essere valutata in modo definitivo. Anche in questo caso, ci sono rapporti individuali positivi.
Infertilità maschile
Il 98% degli uomini con fibrosi cistica sono infertili a causa dell’aplasia bilaterale congenita dei vasi deferenti (CBAVD) che si verifica in fase prenatale e porta all’azoospermia. Poiché la produzione di spermatozoi è solitamente preservata, gli spermatozoi possono essere ottenuti dal testicolo o dall’epididimo e utilizzati per l’inseminazione intrauterina (IUI), la fecondazione in vitro (FIV) o l’iniezione intracitoplasmatica di spermatozoi (ICSI).
Aspetti ginecologici nei pazienti con FC
Nei pazienti con fibrosi cistica, c’è solo una fertilità leggermente ridotta, cioè dovuta all’aumento della viscosità della secrezione cervicale. Numerosi rapporti di casi suggeriscono che la normalizzazione della viscosità attraverso il trattamento con modulatori CFTR porta a un miglioramento della fertilità.
L’opportunità di continuare la terapia con un modulatore CFTR in caso di gravidanza deve essere chiarita in ogni singolo caso. I modulatori CFTR attraversano la barriera placentare, ma non sono stati descritti effetti negativi sul bambino. Anche gli esperimenti sugli animali non hanno mostrato alcuna evidenza di danni.
Letteratura:
- Ramsey BW, et al: Un potenziatore CFTR nei pazienti con fibrosi cistica e la mutazione G551D. N Engl J Med 2011; 365(18): 1663-1672.
- Volkova N, et al: Progressione della malattia nei pazienti con fibrosi cistica trattati con ivacaftor: dati dai registri nazionali di Stati Uniti e Regno Unito. JCF 2020; 10: 68-70.
- Wainwright CE, et al: Tezacaftor-Ivacaftor nei pazienti con fibrosi cistica omozigote per Phe508del CFTR. N Engl J Med 2015; 373(3): 220-231.
- Taylor-Cousar JL, et al: Tezacaftor-Ivacaftor nei pazienti con fibrosi cistica omozigote per Phe508del. N Engl J Med 2017; 377(21): 2013-2023.
- Rowe SM, et al: Tezacaftor-ivacaftor negli eterozigoti a funzione residua con fibrosi cistica. N Engl J Med 2017; 377(21): 2024-2035.
- Middleton et al. Elexacaftor-Tezacaftor-Ivacftor per la fibrosi cistica con un singolo allele Phe508del. N Engl J Med 2019; 381(19): 1809-1819.
- Yang C, Montgomery M.: Dornase alfo per la fibrosi cistica. Cochrane Database Syst Rev 2021; 3(3): CD001127; doi: 10.1002/14651858.CD001127.pub5.
- Wark P, McDonald VM: Salina ipertonica nebulizzata per la fibrosi cistica. Cochrane Database Syst Rev 2018; 9(9): CD001506; doi: 10.1002/14651858.CD001506.pub4.
- Beswick DM, et al: Impatto della terapia con il regolatore della conduttanza transmembrana della fibrosi cistica sulla rinosinusite cronica e sullo stato di salute: analisi CT Deep Learning e risultati riferiti dai pazienti. Ann Am Thorac Soc 2022; 19(1): 12-19.
- Hewer SCL, et al: Strategie antibiotiche per eradicare lo Pseudomonas aeruginosa nelle persone con fibrosi cistica: Cochrane Database Syst Rev 2017; 4(4): CD004197.
InFo PNEUMOLOGIA & ALLERGOLOGIA 2022; 4(4): 14-18









