L’introduzione di nuovi farmaci ha migliorato costantemente la prognosi dei pazienti con mieloma multiplo negli ultimi anni. Anche la diagnostica è stata ampliata, in particolare attraverso le analisi citogenetiche, che consentono una stratificazione del rischio più precisa. Tuttavia, il tasso di sopravvivenza a 5 anni nello stadio III è solo del 40%, quindi c’è ancora spazio per l’innovazione.
Come linfoma a cellule B con proliferazione monoclonale di plasmacellule nel midollo osseo, il mieloma multiplo (MM) è una malattia estremamente eterogenea. Mentre circa un quarto delle persone colpite è asintomatico al momento della diagnosi, esistono anche decorsi acuti con rapida distruzione ossea, disfunzione renale pronunciata, affaticamento, ipercalcemia e tendenza alle infezioni [1]. Con l’invecchiamento della popolazione, il numero di nuovi casi aumenta, con la maggior parte dei casi che si verificano tra i 70 e gli 80 anni. Il mieloma multiplo è ancora una malattia rara, con un’incidenza di circa 6 casi per 100.000 abitanti negli uomini e 4 casi per 100.000 abitanti nelle donne, ma si può prevedere un aumento del numero di casi di un terzo entro il 2040, a causa dello spostamento delle strutture di età da sole [2,3]. I requisiti per una gestione efficiente con opzioni terapeutiche efficaci e una diagnostica orientata al bersaglio continueranno quindi ad aumentare in futuro.
Uno sguardo alla fisiopatologia
Nel mieloma multiplo, le plasmacellule maligne producono immunoglobuline monoclonali complete o incomplete. Queste possono essere rilevate come catene leggere clonalmente aumentate nel siero e nell’urina e portano anche il nome di “paraproteine”. Nell’elettroforesi delle proteine del siero, appaiono sotto forma del famoso “gradiente M”. L’elevata concentrazione di queste immunoglobuline può causare vari sintomi, come l’amiloidosi AL, dovuta alla deposizione di proteine mal ripiegate. Inoltre, può verificarsi una sindrome da iperviscosità e, molto più frequentemente, un deterioramento della funzione renale. Questo perché le catene leggere aumentate vengono filtrate a livello glomerulare e spesso non possono essere riassorbite completamente nei tubuli. Questo porta all’escrezione nelle urine, la cosiddetta “proteinuria di Bence-Jones”. Inoltre, le paraproteine si accumulano nei glomeruli e precipitano nel tubulo distale legandosi alla proteina Tamm-Horsfall prodotta dalle cellule epiteliali tubulari. Risultati: Glomerulosclerosi e nefropatia da calco [4].
Altri sintomi del mieloma multiplo derivano principalmente dallo spostamento della normale emopoiesi e dalla distruzione delle ossa. Questi meccanismi portano a dolori ossei, fratture patologiche, ipercalcemia, anemia e suscettibilità alle infezioni, tra le altre cose [1]. Spesso i disturbi delle persone colpite sono poco specifici, per cui non è raro che passino diversi mesi prima che venga fatta una diagnosi corretta.
La genetica del mieloma multiplo è tanto varia quanto l’aspetto clinico. Le trisomie sono presenti in circa il 40% dei pazienti. Sono comuni anche le traslocazioni che coinvolgono il locus della catena pesante dell’immunoglobulina (IgH) sul cromosoma 14q32. Insieme alle trisomie, queste appartengono alle alterazioni genetiche primarie e possono essere rilevate già nelle fasi preliminari della malattia [5]. Con il progredire della malattia, si aggiungono aberrazioni genetiche secondarie, come la del(1p) o le mutazioni RAS, che modellano la clinica, la risposta alla terapia e la prognosi [3,6].
La conclusione è che l’eziologia del mieloma multiplo non è chiara. Un cluster familiare è raro e i fattori di rischio rimangono poco chiari [3]. La malattia di solito si sviluppa da una gammopatia monoclonale di significato sconosciuto (MGUS) [7]. Se è presente questo stadio preliminare, il rischio di progressione verso il mieloma multiplo o un’altra malattia linfoproliferativa che richiede un trattamento è di circa l’1% all’anno [8]. Tuttavia, la diagnosi precoce dei precursori non è stata stabilita, poiché non ha ancora portato a un miglioramento significativo della prognosi [3].
Diagnostica in transizione
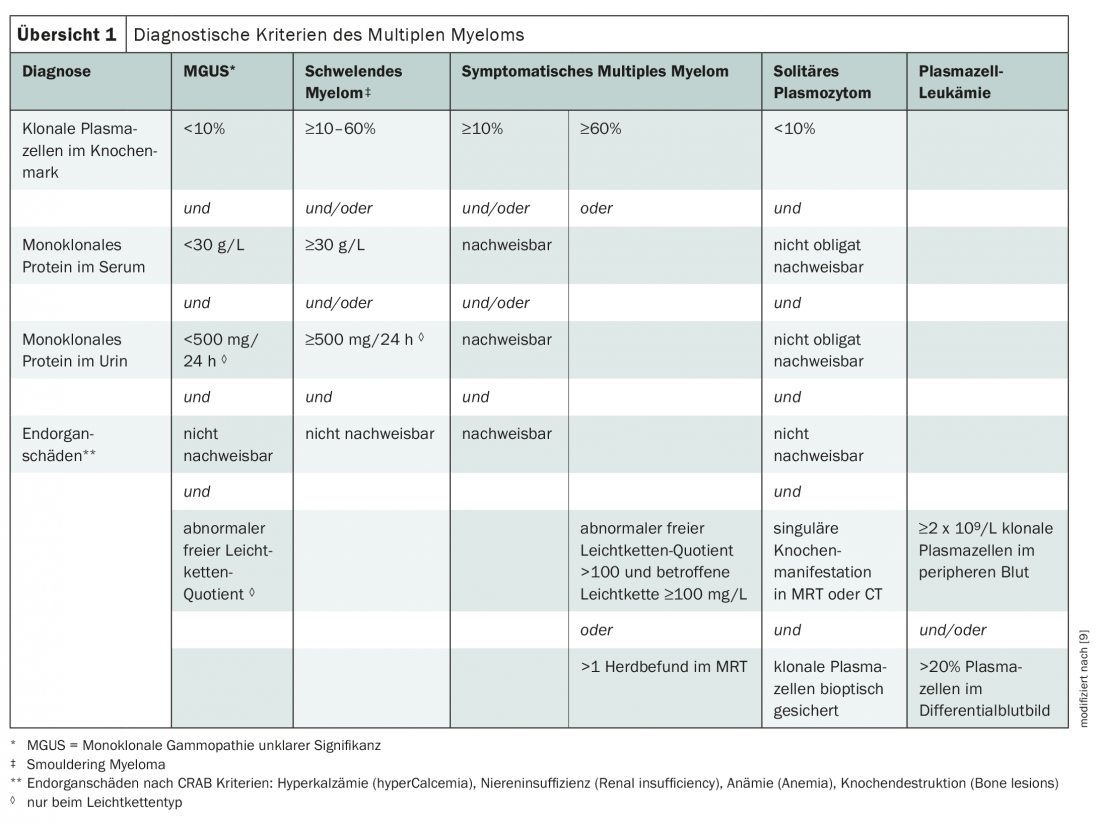
Attualmente, i criteri dell’International Myeloma Working Group sono utilizzati principalmente per la diagnosi di mieloma multiplo (panoramica 1) [3,9]. La malattia è classificata in base al tipo di paraproteina, con i mielomi IgG e IgA che sono i più comuni. Se si formano solo immunoglobuline incomplete, cioè catene leggere, si parla di “mieloma a catena leggera”. Questi rappresentano circa un quinto dei casi [3].
Oltre all’anamnesi, all’esame fisico, all’emocromo e al laboratorio, fanno parte della diagnostica iniziale anche una TAC a basso dosaggio del corpo intero e una puntura del midollo osseo. La diagnostica per immagini viene utilizzata per rilevare l’osteolisi e l’osteopenia [3]. Se necessario, questo esame può essere ampliato da una risonanza magnetica per una più precisa differenziazione dei focolai ossei e per la diagnosi di manifestazioni extramidollari. Questo può essere particolarmente utile per differenziare il mieloma da quello in via di estinzione [10]. La scansione FDG-PET può anche fornire informazioni sui focolai extramidollari e sulla risposta terapeutica, ma attualmente non fa parte delle indagini standard [11]. Una risonanza magnetica di regioni specifiche deve essere ordinata al più tardi prima di iniziare la terapia, se si sospetta un coinvolgimento extramidollare o se sono presenti sintomi neurologici. Anche l’ecocardiografia è indicata se si sospetta un’amiloidosi cardiaca [3].
Sempre più spesso, la diagnostica genetica svolge un ruolo importante nella gestione del mieloma multiplo. Le analisi su del(17p), t(4; 14) e t(14; 16) corrispondono oggi al work-up minimo [3]. Questi cambiamenti genetici sono associati a una prognosi significativamente peggiore. Altre aberrazioni e anche i punteggi prognostici basati sull’espressione genica sono rilevanti dal punto di vista prognostico, ma attualmente non sono (ancora) predittivi per terapie specifiche [6,12].
Classificazione per valutare la prognosi
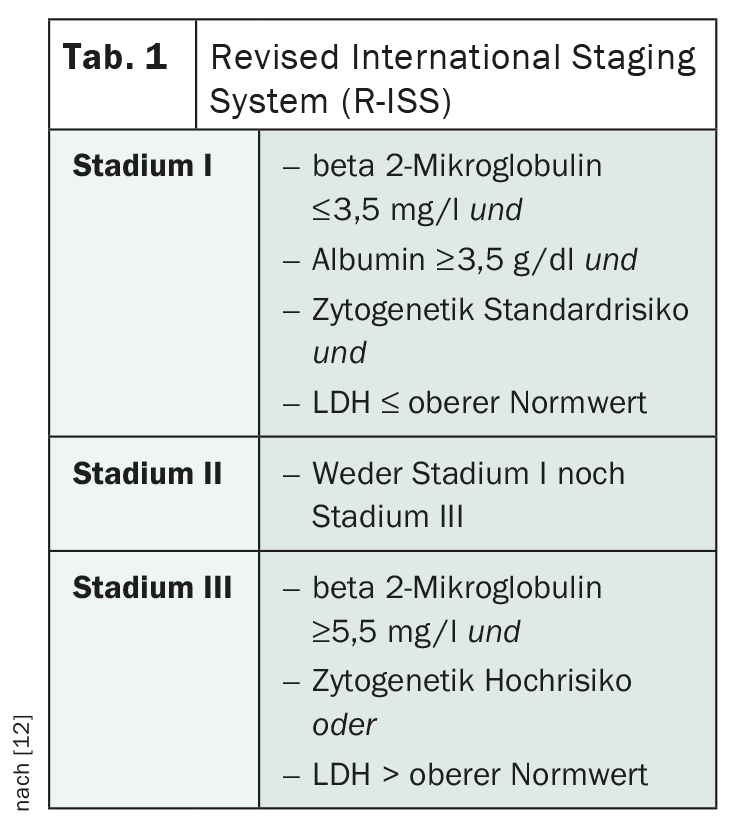
La stadiazione del mieloma multiplo gioca un ruolo importante nella stima della prognosi e nella valutazione della terapia più appropriata. La classificazione secondo Salmon e Durie, in uso da molto tempo, è ormai obsoleta. Al suo posto viene utilizzato il Revised International Staging System (R-ISS ) dell’International Myeloma Working Group ( IMWG) (Tab. 1) [12]. In base a questa stadiazione, i soggetti colpiti vengono suddivisi in tre gruppi prognostici, tenendo conto dell’albumina sierica, della beta 2-microglobulina, dell’LDH e delle aberrazioni citogenetiche.
Anche la presenza di malattia minima residua (MRD) dopo la terapia è prognosticamente significativa. È presente nella grande maggioranza dei pazienti anche dopo aver raggiunto una remissione completa secondo i criteri attuali e si correla con un esito peggiore [13]. Poiché le analisi della MRD non hanno ancora un valore predittivo per un’ulteriore terapia, attualmente non sono uno standard negli esami di follow-up [3].
La terapia di prima linea in sintesi
Oltre ai decorsi sintomatici e ai cosiddetti criteri CRAB (ipercalcemia – C, insufficienza renale – R, anemia – A e coinvolgimento osseo – B), esistono anche altri parametri radiologici e sierologici che, secondo i criteri IMWG, rappresentano indicazioni per l’inizio della terapia. Questi includono l’evidenza di biomarcatori che definiscono il mieloma, un contenuto di plasmacellule clonali nel midollo osseo di >60%, un quoziente di catene leggere libere nel siero di >100 (catene leggere colpite/non colpite) e lesioni focali di oltre 1 cm alla risonanza magnetica [3].
Se esiste un’indicazione per la terapia, si distingue principalmente tra i pazienti che sono adatti al trapianto di cellule staminali e quelli che devono essere trattati senza terapia ad alto dosaggio a causa di comorbidità. Se il trapianto non è un’opzione, nel trattamento farmacologico si possono utilizzare varie combinazioni di due o tre farmaci. I pazienti affetti da insufficienza renale, elevata attività di malattia e citogenetica sfavorevole devono preferibilmente ricevere una terapia con bortezomib come componente [3]. Altri agenti utilizzati sono ciclofosfamide, melfalan, lenalidomide e steroidi. Purtroppo, solo alcuni degli schemi comuni sono stati confrontati direttamente. Nel complesso, le combinazioni di tre sostanze che utilizzano un inibitore del proteasoma, un immunomodulatore e uno steroide sembrano essere superiori alle combinazioni doppie. Se le condizioni generali sono buone, sono quindi preferibili [3].
Se è disponibile il trapianto di cellule staminali autologhe, è il trattamento di scelta nella prima linea di terapia. Finora, nessun nuovo farmaco è stato in grado di eguagliare il trapianto in termini di tasso di remissione, profondità della risposta e sopravvivenza libera da progressione [14,15]. Gli effetti avversi della terapia ad alto dosaggio sono un fattore limitante per l’indicazione del trapianto di cellule staminali autologhe. Ciò richiede una buona funzionalità degli organi e l’assenza di comorbidità significative [16]. Solo pochi anni fa, veniva raccomandata una terapia di induzione contenente vincristina-antraciclina. Da allora, queste terapie sono state sostituite da terapie combinate con i nuovi farmaci talidomide, bortezomib e lenalidomide, che portano a tassi di risposta significativamente migliori. Anche in questo caso, i dati sul confronto diretto delle diverse terapie combinate sono purtroppo limitati; le remissioni complete si ottengono nel 20-40% dei casi [3]. Attualmente, la scelta della terapia di induzione si basa principalmente su fattori individuali del paziente, effetti collaterali e disponibilità di farmaci.
Il trapianto di cellule staminali dopo la terapia di induzione può essere eseguito come trapianto singolo o in tandem. In quest’ultimo caso, si esegue un secondo trapianto autologo entro sei mesi. Questo porta a un prolungamento della sopravvivenza libera da progressione e nei pazienti in stadio R-ISS III è stata osservata una sopravvivenza globale più lunga oltre a una sopravvivenza libera da progressione più lunga [17]. Tuttavia, si deve tenere conto anche della maggiore tossicità di una seconda terapia ad alto dosaggio. Attualmente, il trapianto tandem è raccomandato per i pazienti del gruppo R-ISS III e per quelli con citogenetica ad alto rischio, tenendo conto di varie analisi di sottogruppo e a lungo termine [3]. Naturalmente, è necessario garantire la presenza di un numero sufficiente di cellule staminali autologhe conservate. Oggi, il melfalan 200 mg/m2 è utilizzato per la terapia ad alte dosi [18]. I regimi di condizionamento alternativi che utilizzano la ciclofosfamide e/o l’irradiazione totale del corpo non sono più raccomandati a causa della loro maggiore tossicità. Anche la terapia ottimale ad alte dosi è attualmente ancora oggetto di diversi studi; una somministrazione aggiuntiva di busulfano non ha portato a un prolungamento della sopravvivenza globale. Purtroppo, l’aggiunta di bortezomib non si è dimostrata efficace [19]. Non ci sono dati conclusivi sull’attuazione della terapia di consolidamento dopo il trapianto di cellule staminali autologhe; questa potrebbe essere particolarmente utile nei pazienti con risposta subottimale dopo il trapianto o con citogenetica ad alto rischio [20].
Le raccomandazioni per la terapia di mantenimento sono più chiare. Dopo anni in cui non era raccomandata, oggi è una parte importante dello standard terapeutico, grazie ai nuovi farmaci. Quindi, il bortezomib o la lenalidomide vengono utilizzati nel gruppo ad alto rischio e la lenalidomide nel rischio standard. E questo è anche il caso della risposta completa [3,19,21].
Un fattore terapeutico importante da non dimenticare, indipendentemente dallo stato del trapianto, è l’osteoprotezione. Soprattutto nei casi di coinvolgimento osseo e durante la terapia con glucocorticoidi, è necessario utilizzare bifosfonati o – soprattutto nei casi di insufficienza renale – denosumab per prevenire al meglio la perdita ossea. Lo zolendronato è considerato il bifosfonato di prima scelta per il mieloma multiplo [3].
E in caso di progressione o recidiva?
Le progressioni e le ricadute dopo la terapia di prima linea rappresentano una sfida importante nel mieloma multiplo. Anche perché la popolazione di pazienti è ancora più disomogenea con le linee di terapia successive. Le persone colpite hanno ricevuto trattamenti diversi e hanno avuto esperienze diverse. La scelta dei farmaci si basa, tra l’altro, sull’efficacia e sulla tollerabilità del trattamento di prima linea. Quindi, se l’esperienza di prima linea è buona, si può utilizzare un farmaco della stessa classe di sostanze nel trattamento di seconda linea. In caso di bassa efficacia o scarsa tollerabilità, tuttavia, è indicato un cambio di classe farmacologica. Attualmente c’è un’abbondanza di nuovi farmaci e combinazioni di farmaci. A seconda della presentazione clinica, delle terapie precedenti e delle comorbidità, si può quindi selezionare un’ampia gamma di sostanze e sequenze [3].
Mentre le ricadute precoci e tardive vengono trattate preferibilmente con il trapianto di cellule staminali, esistono vari regimi terapeutici per tutte le altre ricadute e per le controindicazioni al trapianto. Sono a base di inibitori del proteasoma o di immunomodulatori e sono composti da due o tre sostanze attive (panoramica 2) . Qui viene utilizzato anche l’anticorpo anti-CD38 daratumumab. In generale, le combinazioni triple sono più efficaci di quelle doppie, ma bisogna tenere conto anche delle maggiori tossicità [3,19].

Con la crescente ricerca di nuovi agenti attivi per la terapia del mieloma multiplo, negli ultimi anni sono stati raggiunti alcuni successi, in particolare nella terapia di mantenimento e nelle linee di trattamento avanzate. Tuttavia, la prognosi nel gruppo ad alto rischio e nella malattia ricorrente è ancora oggi sfavorevole. Grazie all’analisi citogenetica sempre più diffusa, è già possibile una migliore stratificazione del rischio. Questo fa sperare che in futuro saranno disponibili non solo opzioni diagnostiche, ma anche terapeutiche potenzialmente più mirate.
Letteratura:
- Kyle RA, et al: Revisione di 1027 pazienti con mieloma multiplo di nuova diagnosi. Mayo Clin Proc. 2003; 78(1): 21-33.
- NICER: Statistiche nazionali sull’incidenza del cancro. www.nicer.org/en/statistics-atlas/cancer-incidence (ultimo accesso 27.02.2021)
- Wörmann B, et al: Mieloma multiplo. Linea guida dell’Oncopedia. www.onkopedia.com/de/onkopedia/guidelines/multiples-myelom/@@guideline/html/index.html (ultimo accesso 27.02.2021)
- Dimopoulos MA, et al: Patogenesi e trattamento dell’insufficienza renale nel mieloma multiplo. Leucemia. 2008; 22(8): 1485-1493.
- Mikulasova A, et al.: Lo spettro di mutazioni somatiche nella gammopatia monoclonale di significato indeterminato indica un paesaggio genomico meno complesso di quello del mieloma multiplo. Ematologica. 2017; 102(9): 1617-1625.
- Bergsagel PL, Chesi MV: Classificazione molecolare e stratificazione del rischio del mieloma. Hematol Oncol. 2013; 31 Suppl 1(0 1): 38-41.
- Landgren O, et al.: La gammopatia monoclonale di significato indeterminato (MGUS) precede costantemente il mieloma multiplo: uno studio prospettico. Sangue. 2009; 113(22): 5412-5417.
- Dispenzieri A, et al: Prevalenza e rischio di progressione della gammopatia monoclonale a catena leggera di significato indeterminato: uno studio di coorte retrospettivo basato sulla popolazione. Lancet. 2010; 375(9727): 1721-1728.
- Rajkumar SV, et al: Criteri aggiornati dell’International Myeloma Working Group per la diagnosi di mieloma multiplo. Lancet Oncol. 2014; 15(12): e538-548.
- Dimopoulos MA, et al: Ruolo della risonanza magnetica nella gestione dei pazienti con mieloma multiplo: una dichiarazione di consenso. J Clin Oncol. 2015; 33(6): 657-664.
- Cavo M, et al: Ruolo della (18)F-FDG PET/CT nella diagnosi e nella gestione del mieloma multiplo e di altri disturbi delle plasmacellule: una dichiarazione di consenso dell’International Myeloma Working Group. Lancet Oncol. 2017; 18(4): e206-e17.
- Palumbo A, et al: Sistema di stadiazione internazionale rivisto per il mieloma multiplo: un rapporto del Gruppo di lavoro internazionale sul mieloma. J Clin Oncol. 2015; 33(26): 2863-2869.
- Munshi NC, et al: Associazione della malattia minima residua con esiti di sopravvivenza superiori nei pazienti con mieloma multiplo: una meta-analisi. JAMA Oncol. 2017; 3(1): 28-35.
- Barlogie B, et al.: Superiorità del trapianto autologo in tandem rispetto alla terapia standard per il mieloma multiplo non trattato in precedenza. Sangue. 1997; 89(3): 789-793.
- Gay F, et al: Chemioterapia più lenalidomide rispetto al trapianto autologo, seguito da lenalidomide più prednisone rispetto al mantenimento di lenalidomide, nei pazienti con mieloma multiplo: uno studio randomizzato, multicentrico, di fase 3. Lancet Oncol. 2015; 16(16): 1617-1629.
- Merz M, et al: Sopravvivenza dei pazienti anziani con mieloma multiplo – effetto del trapianto di cellule staminali autologhe in anticipo. Eur J Cancer. 2016; 62: 1-8.
- Cavo M, et al: Il doppio trapianto di cellule staminali autologhe prolunga significativamente la sopravvivenza libera da progressione e la sopravvivenza complessiva rispetto all’autotrapianto singolo nel mieloma multiplo di nuova diagnosi: un’analisi dello studio di fase 3 EMN02/HO95. Sangue. 2017; 130 (supplemento 1): 401.
- Giralt S: 200 mg/m(2) di melfalan – il gold standard per il mieloma multiplo. Nat Rev Clin Oncol. 2010; 7: 490-491.
- Dimopoulos MA, et al: Mieloma multiplo: Linee guida di pratica clinica EHA-ESMO per la diagnosi, il trattamento e il follow-up. Annali di Oncologia. 2021; 32(3): 309-322.
- Einsele H, et al: Il consolidamento adattato alla risposta con bortezomib dopo ASCT migliora la sopravvivenza libera da progressione nel mieloma multiplo di nuova diagnosi. Leucemia. 2017; 31(6): 1463-1466.
- McCarthy PL, et al: Mantenimento della lenalidomide dopo il trapianto autologo di cellule staminali nel mieloma multiplo di nuova diagnosi: una meta-analisi. J Clin Oncol. 2017; 35(29): 3279-3289.
InFo ONCOLOGIA & EMATOLOGIA 2021; 9(2): 22-26