Con l’invecchiamento della società, anche le malattie MDS sono in aumento. Tuttavia, gli sviluppi nella diagnostica molecolare e le nuove terapie cambieranno anche la gestione dei pazienti anziani. Un approccio multidisciplinare e lo scambio sono importanti.
Le sindromi mielodisplastiche (MDS) sono diagnosticate come malattie degli anziani, principalmente nei pazienti >70 anni. In Svizzera, con un’incidenza di 2-3/100.000 anni-paziente, si possono prevedere poco più di 300 nuovi casi all’anno. Attualmente si stima che nel nostro Paese vi siano circa 1600 pazienti affetti da MDS [1]. Le opzioni terapeutiche sono rimaste sostanzialmente invariate negli ultimi anni, con il trapianto di cellule staminali allogeniche come trattamento curativo possibile solo per pochi pazienti, e varie opzioni per migliorare le citopenie nelle restanti situazioni palliative [2–5]. Al contrario, il rapido sviluppo del sequenziamento di “prossima generazione” (NGS) ha portato anche a nuove scoperte rilevanti nella routine clinica dell’ematologia [6,7] che svolgono un ruolo per i pazienti con MDS per quanto riguarda la diagnosi e la valutazione della prognosi. Sulla base della classificazione aggiornata dell’OMS del 2016, di seguito verranno presentati gli sviluppi e i concetti più importanti.
Revisione della Classificazione OMS 2016
Anche nell’era della biologia molecolare, la valutazione morfologica degli strisci di sangue periferico e degli aspirati di midollo osseo rimane il fondamento della diagnostica. Fondamentalmente, le forme di MDS con eccesso di blasti si distinguono da quelle senza proliferazione di blasti. Il numero di file di cellule interessate da citopenie e displasie, il rilevamento di sideroblasti ad anello (RS) e i cambiamenti citogenetici tipici sono decisivi per un’ulteriore suddivisione. Una novità della classificazione OMS 2016 è la nomenclatura (Tab. 1) [8,9]. I termini “anemia refrattaria” o “citopenia refrattaria” sono stati abbandonati e il termine “sindrome mielodisplastica” viene ora utilizzato per tutte le entità, integrato dal reperto morfologico centrale. Questo chiarisce alcune incongruenze nella terminologia precedente, come il termine “anemia refrattaria” per le forme di MDS con eccesso di blasti, spesso accompagnate da pancitopenia.

La citogenetica metafasica convenzionale rimane il secondo pilastro indispensabile della diagnostica delle MDS. Nei pazienti con citopenie senza proliferazione di blasti e senza displasie (significative), la diagnosi può essere fatta rilevando alcune anomalie citogenetiche che definiscono la MDS ( Tab. 2). In questo caso, viene assegnata la diagnosi “MDS non classificabile”. Questa categoria comprende anche i casi con pancitopenia e displasia unilineare o con rilevamento costante di blasti dell’1% nel sangue periferico senza moltiplicazione di blasti nel midollo osseo [8,9].
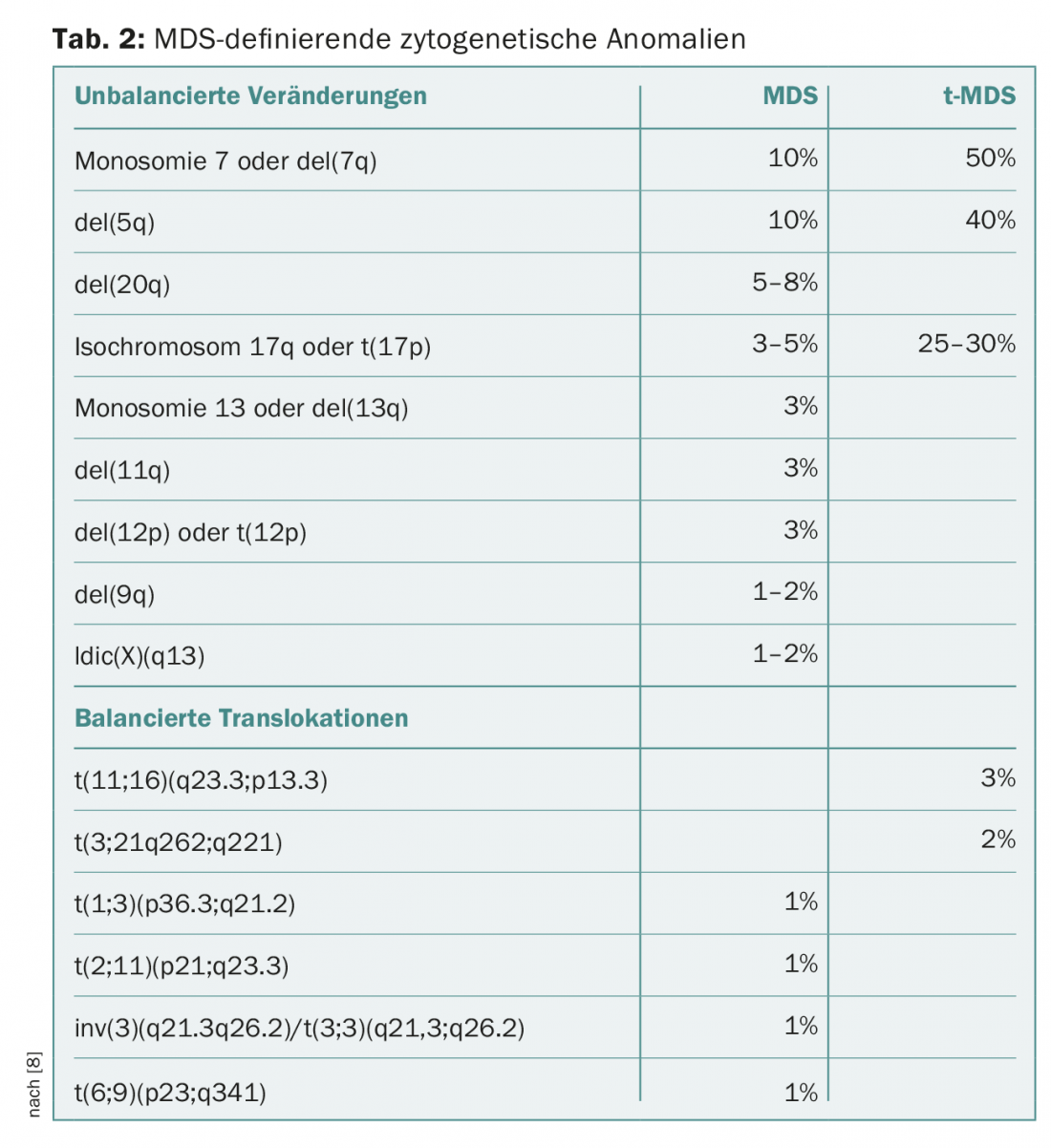
La categoria delle SMD caratterizzate da una delezione sul braccio corto del cromosoma 5 con del(5q) è stata ampliata nella sua definizione. Ora, anche i casi con una seconda anomalia citogenetica possono essere assegnati a questa categoria, tranne nel caso di anomalie aggiuntive nel cromosoma 7, che sono associate a una prognosi significativamente peggiore [8–10].
La citogenetica convenzionale (l’analisi di almeno 20 metafasi è necessaria per un esame conclusivo) può essere integrata da altri metodi in situazioni selezionate, ad esempio nel caso di un sospetto morfologicamente elevato di MDS del(5q) ma con cariotipo normale. Possono essere utilizzati pannelli FISH focalizzati sulle anomalie cromosomiche tipiche della MDS o microarray di ibridazione genomica comparativa (array-CGH) [11]. Tuttavia, questi due metodi non offrono una sostituzione a priori della citogenetica convenzionale. In particolare, il significato prognostico delle anomalie che possono essere rilevate solo nell’array CGH non è stato ancora chiarito con certezza.
Significato diagnostico del sequenziamento di “prossima generazione” nelle citopenie non chiare
Negli ultimi anni, sono state identificate numerose mutazioni nei geni che sono associate allo sviluppo di MDS e alla progressione in AML. Lo spettro dei “geni driver” mutati è vario e spesso include componenti della modificazione epigenetica del DNA e degli istoni o il macchinario di splicing che modifica l’RNA (“spliceosoma”). Possono essere interessati anche i componenti della regolazione del ciclo cellulare, i complessi di coesina, i fattori di trascrizione o i componenti della trasduzione del segnale intracellulare (Tab. 3) [2,12–18].
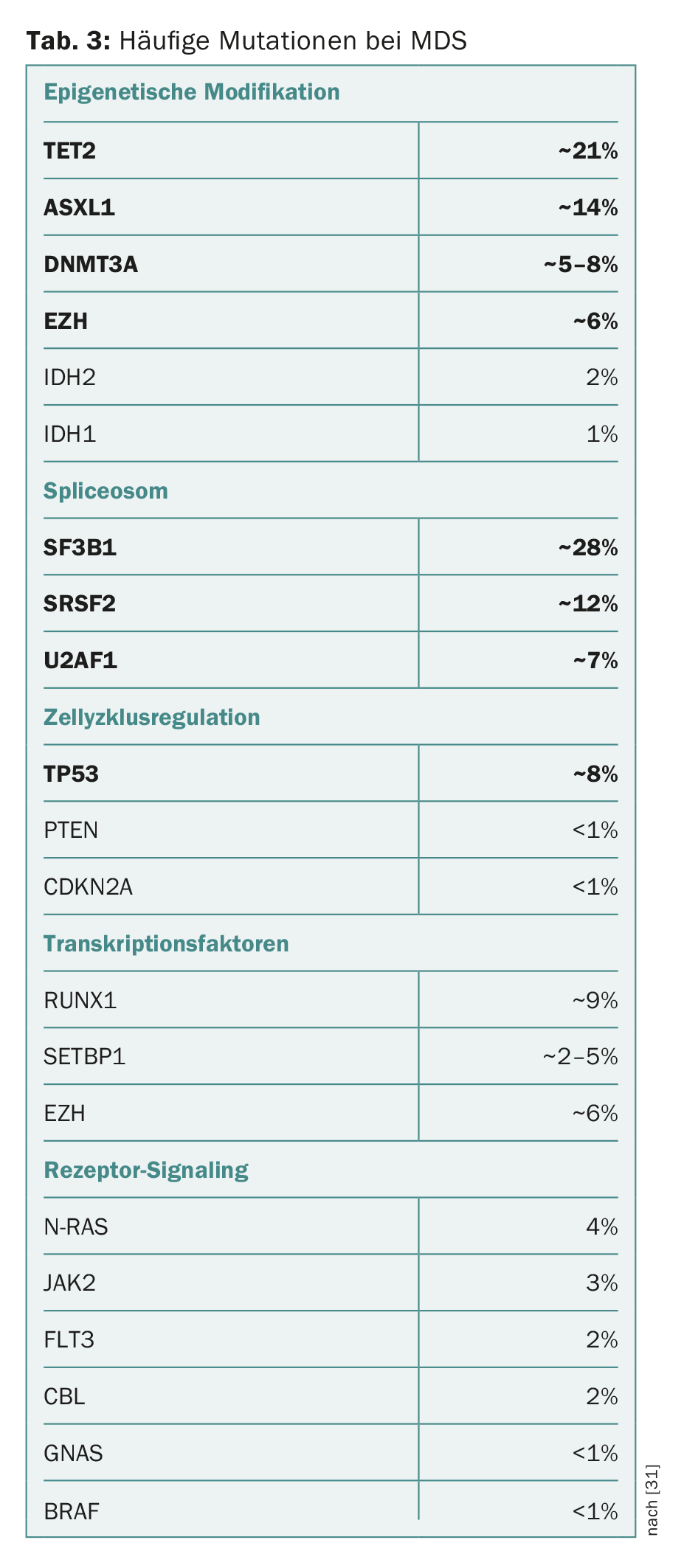
Da un lato, va sottolineato che la maggior parte delle mutazioni dei geni driver riscontrate nella SMD si verificano anche in altre neoplasie mieloidi, anche se con frequenze o combinazioni diverse. Inoltre, diversi studi hanno dimostrato che le mutazioni tipiche delle neoplasie mieloidi possono verificarsi anche in individui ematologicamente sani. L’incidenza aumenta bruscamente con l’età, dal 10% nei 60enni al 15-20% negli >80enni (ma <1% nei <40enni). Questo fenomeno è stato definito “Ematopoiesi clonale a potenziale indeterminato” (CHIP) [19,20]. Simile alla gammopatia monoclonale di significato sconosciuto (MGUS) e alla linfocitosi monoclonale a cellule B (MBL), è una condizione precancerosa facoltativa che può progredire in una malattia ematologica maligna a un tasso di circa l’1% all’anno. Inoltre, i pazienti con CHIP presentano anche una maggiore morbilità cardiovascolare [21].
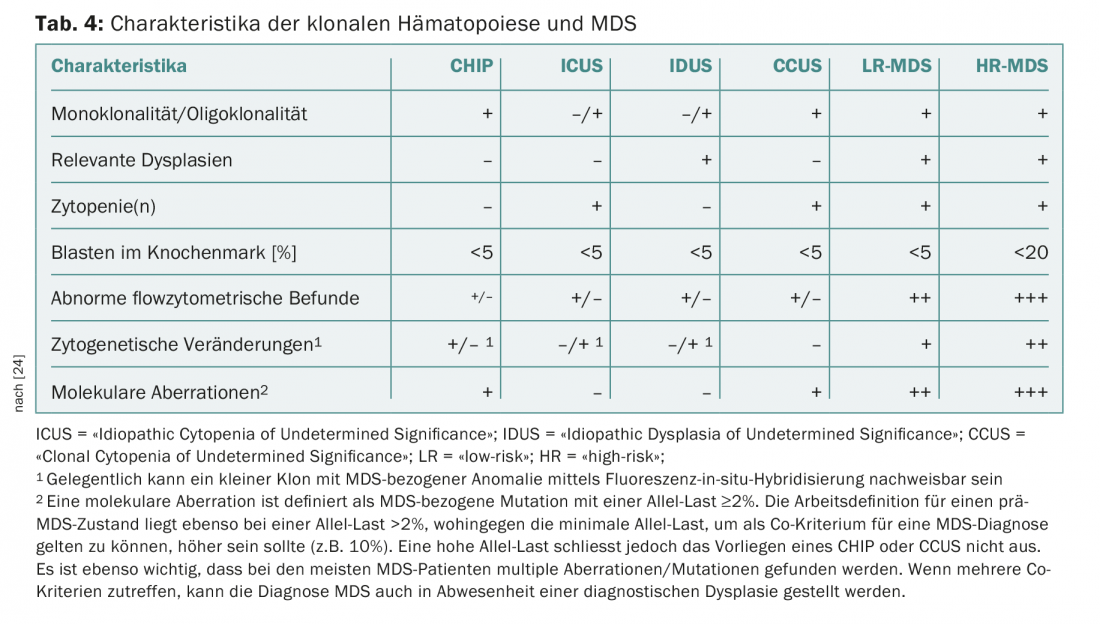
Un problema ricorrente nella pratica e nella clinica sono i pazienti con citopenie persistenti per le quali non si riesce a trovare una causa certa. In assenza di displasia o di anomalie citogenetiche che definiscono la MDS, queste sono state finora raggruppate sotto il termine di “citopenia idiopatica di significato indeterminato” (ICUS) [22]. Il rilevamento di mutazioni ricorrenti può aiutare a distinguere le citopenie reattive da quelle clonali. L’interpretazione del rilevamento di una mutazione dipende dalla dimensione del clone (misurata come “frequenza allelica variante”, VAF) e dal numero di mutazioni rilevate. Se in una costellazione ICUS si trova una mutazione ricorrente con un VAF superiore al 2%, si parla di “citopenia clonale di importanza indeterminata” (CCUS). Tra i pazienti con CCUS, quelli con almeno due mutazioni con un VAF >10% sono ad alto rischio di sviluppare una neoplasia ematologica nei cinque anni successivi [23]. I criteri presentati di recente da un gruppo di esperti internazionali per distinguere CHIP, ICUS, CCUS dalla MDS manifesta [24] sono riassunti nella tabella 4. Questo consenso definisce anche nuovi criteri minori legati alla MDS, che possono essere utilizzati per una diagnosi provvisoria di MDS in situazioni non conclusive (Tab. 5).

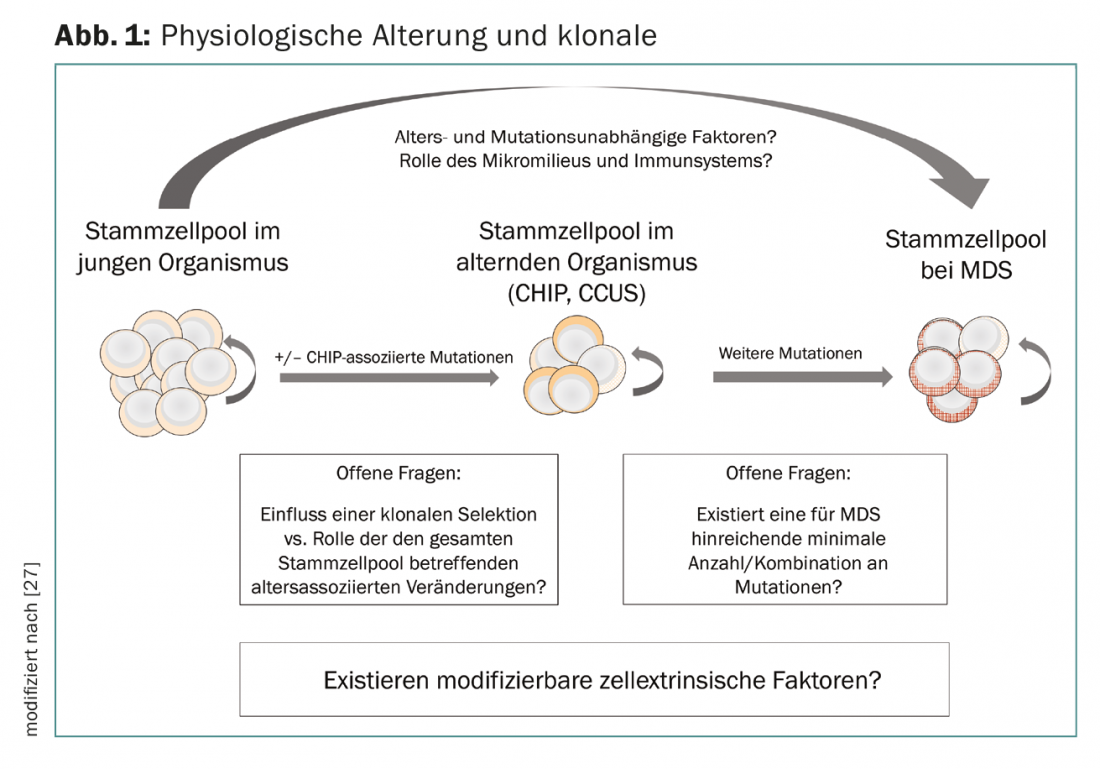
L’accumulo sequenziale di alterazioni genetiche nelle CSE è stato a lungo postulato come un correlato patogenetico dell’evoluzione clonale, e il concetto di ematopoiesi clonale dimostra una sovrapposizione tra le alterazioni dell’ematopoiesi con l’età e la patogenesi delle neoplasie mieloidi. Tuttavia, rimane poco chiaro quali fattori siano responsabili della transizione da CHIP e CCUS a neoplasia manifesta. L’evoluzione clonale sembra essere causata non solo da meccanismi intrinseci alle cellule (mutazioni nelle cellule staminali ematopoietiche), ma anche da meccanismi estrinseci alle cellule. A questo proposito, il microambiente del midollo osseo e i componenti del sistema immunitario innato e acquisito svolgono un ruolo. Lo “stress immunologico” spiega probabilmente anche l’associazione con malattie o fenomeni infiammatori e immunologici concomitanti, alcuni dei quali non possono essere ulteriormente classificati, che possono verificarsi nei pazienti con MDS [25–28]. La perdita del controllo immunologico del tumore e i cambiamenti favorevoli nella nicchia del midollo osseo sono attualmente al centro della ricerca di base. In futuro, questo potrebbe portare a nuovi approcci terapeutici che potrebbero essere utilizzati precocemente nello sviluppo delle neoplasie mieloidi (Fig. 1).
A causa della sovrapposizione dello spettro di mutazioni tra CHIP, CCUS e MDS, le analisi delle mutazioni non sono state volutamente incluse nell’attuale classificazione OMS. Un’eccezione è rappresentata dalle mutazioni nel gene driver del componente dello spliceosoma SF3B1, che sono fortemente associate a un fenotipo sideroblastico ad anello [29]. Secondo l’OMS 2016, nel caso di una mutazione SF3B1, il rilevamento del 5% di RS è sufficiente per la classificazione nel gruppo di MDS con RS, invece del 15% altrimenti richiesto. I pazienti mutati in SF3B1 hanno una prognosi molto buona, con una bassa probabilità di progressione in AML. Poiché i pazienti con displasia multilinea e RS beneficiano anche dell’influenza prognostica di una mutazione SF3B1, l’entità MDS con displasia multilinea e RS è stata reintrodotta nella classificazione del 2016.
Ulteriore significato prognostico e predittivo delle mutazioni del gene driver
Da un punto di vista prognostico, si possono già nominare alcuni ulteriori scenari clinici in cui le analisi delle mutazioni possono fornire informazioni rilevanti. Circa il 15% dei pazienti con MDS del(5q) presenta una mutazione TP53. Sebbene anche questi rispondano ematologicamente alla lenalidomide, hanno meno probabilità di raggiungere la remissione citogenetica e presentano un rischio più elevato di transizione verso l’AML. Pertanto, se viene rilevata una mutazione TP53 nella MDS del(5q), si possono prendere in considerazione terapie alternative [3,30].
Un altro scenario per la ricerca di mutazioni riguarda il gruppo eterogeneo di pazienti del gruppo “intermedio” secondo l’IPSS-R. A seconda della presenza o dell’assenza di altre caratteristiche di rischio, questi pazienti possono essere trattati secondo le raccomandazioni applicabili ai pazienti a “basso” o “alto rischio” [2–5,30]. Finora, a questo scopo sono disponibili solo i marcatori di rischio convenzionali (ad esempio, elevazione della LDH, fibrosi del midollo osseo >di grado 2 secondo l’OMS), oltre all’esame isolato della citogenetica (costellazione ad alto rischio?). Diverse mutazioni ricorrenti sono associate a un rischio significativamente maggiore e giustificano un upstaging prognostico alla categoria “ad alto rischio” e, in pazienti selezionati, il trapianto di cellule staminali allogeniche [15]. La ricerca di mutazioni TP53, ASXL1, RUNX1, EZH2 ed ETV6 nei pazienti che si qualificano per la terapia intensiva è quindi esplicitamente raccomandata nelle attuali linee guida [4,15]. Un progetto internazionale sponsorizzato dalla MDS Foundation sta attualmente cercando di sviluppare un “IPSS-R molecolare” integrato dallo stato di mutazione.
I dati clinicamente utili sul valore predittivo del profilo di mutazione nelle SMD sono attualmente limitati. Sta emergendo una rilevanza per la stratificazione in relazione alle terapie mirate per il futuro. Luspatercept (ACE-356) è un inibitore della superfamiglia TGF-β [10], che ha dimostrato un alto tasso di risposta eritroide nei pazienti MDS refrattari all’EPO con RS e/o mutazioni in SF3B1. Inoltre, gli inibitori dello spliceosoma (H3B-8800) sono attualmente in fase di studio nella LAM e nella SMD che, a causa dell’aplo-insufficienza, eliminano preferenzialmente i cloni con mutazioni nei geni driver dello spliceosoma (effetto ciclope). Inoltre, le sostanze midostaurin (mutazioni FLT3), enasidenib (mutazioni IDH2) e ivosidenib (mutazioni IDH1), già approvate per la LAM, sono attualmente in fase di sviluppo clinico anche per le SMD ad alto rischio con un profilo di mutazione corrispondente, sia come sostanza singola che in combinazione con la terapia ipometilante (HMT) o la chemioterapia standard.
Messaggi da portare a casa
- A causa dell’invecchiamento della nostra società, è prevedibile un aumento significativo della malattia MDS.
- Gli sviluppi nella diagnostica molecolare e le nuove opzioni terapeutiche cambieranno le strategie di trattamento anche per i pazienti più anziani.
- La gestione multidisciplinare è un prerequisito importante e pone nuove sfide ai sistemi sanitari. Ciò riguarda non solo l’assistenza adeguata nella pratica clinica di routine, ma anche la conduzione di studi clinici, che richiedono un alto grado di cooperazione e coordinamento nel caso di malattie rare nell’era della medicina personalizzata.
- In risposta a queste sfide, il Gruppo di Studio MDS svizzero ha lanciato il Registro/Biobanca MDS svizzero nel 2015 per facilitare lo scambio clinico e scientifico all’interno di una rete internazionale.
Letteratura:
- Bonadies N, et al: Tendenze della classificazione, dell’incidenza, della mortalità e della sopravvivenza dei pazienti con MDS in Svizzera tra il 2001 e il 2012. Epidemiologia del cancro 2017; 46: 85-92.
- Malcovati L, et al: Diagnosi e trattamento delle sindromi mielodisplastiche primarie negli adulti: Raccomandazioni dell’European LeukemiaNet. Sangue 2013; 122(17): 2943-2964.
- Fenaux P, et al: Sindromi mielodisplastiche: Linee guida di pratica clinica ESMO per la diagnosi, il trattamento e il follow-up. Annali di oncologia: rivista ufficiale della Società Europea di Oncologia Medica 2014; 25(Suppl 3): iii57-69.
- Hofmann WK, Platzbecker U, Götze K: Onkopedia-Leitlinie Myelodysplastische Syndrome. Stato marzo 2016.
- Greenberg PL, et al: Sindromi mielodisplastiche, versione 2.2017, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network: JNCCN 2017; 15(1): 60-87.
- Johnsen JM, Nickerson DA, Reiner AP: Sequenziamento massicciamente parallelo: la nuova frontiera della genomica ematologica. Sangue 2013; 122(19): 3268-3275.
- Kuo FC, et al: Le utilità relative del sequenziamento genome-wide, del pannello genico e del singolo gene nella pratica clinica. Sangue 2017; 130(4): 433-439.
- Swerdlow SH, et al. (Eds.): Classificazione OMS dei tumori dei tessuti ematopoietici e linfoidi. Quarta edizione riveduta. Lione: Agenzia Internazionale per la Ricerca sul Cancro 2017.
- Arber DA, et al: La revisione 2016 della classificazione dell’Organizzazione Mondiale della Sanità delle neoplasie mieloidi e della leucemia acuta. Sangue 2016; 127(20): 2391-2405.
- Mies A, Platzbecker U: Aumentare l’efficacia dell’emopoiesi nelle sindromi mielodisplastiche: agenti stimolanti l’eritropoiesi e inibitori della superfamiglia del fattore di crescita trasformanteβ. Seminari di ematologia 2017; 54(3): 141-146.
- Ouahchi I, et al.: L’ibridazione genomica comparativa basata su microarray rivela ulteriori aberrazioni ricorrenti in pazienti adulti valutati per la sindrome mielodisplastica con cariotipo normale. British journal of haematology 2018. DOI: 10.1111/bjh.15068 [Epub ahead of print].
- Kon A, et al.: Mutazioni ricorrenti in più componenti del complesso della coesina nelle neoplasie mieloidi. Genetica della natura 2013; 45(10): 1232-1237.
- Yoshida K, et al.: Frequenti mutazioni di percorso del macchinario di splicing nella mielodisplasia. Natura 2011; 478(7367): 64-69.
- Bejar R, Levine R, Ebert BL: Svelare la fisiopatologia molecolare delle sindromi mielodisplastiche. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2011; 29(5): 504-515.
- Bejar R, et al.: Effetto clinico delle mutazioni puntiformi nelle sindromi mielodisplastiche. The New England journal of medicine 2011; 364(26): 2496-2506.
- Abdel-Wahab O, Figueroa ME: Interpretare la nuova genetica molecolare nelle sindromi mielodisplastiche. Ematologia American Society of Hematology Education Program 2012; 2012: 56-64.
- Leeke B, et al.: Mutazioni della coesina nei tumori maligni mieloidi: meccanismi sottostanti. Ematologia & Oncologia sperimentale 2014; 3: 13.
- Tothova Z, Steensma DP, Ebert BL: Nuove strategie nelle sindromi mielodisplastiche: Applicazione della diagnostica molecolare alla pratica clinica. Clinical cancer research: an official journal of the American Association for Cancer Research 2013; 19(7): 1637-1643.
- Jan M, Ebert BL, Jaiswal S: Ematopoiesi clonale. Seminari di ematologia 2017; 54(1): 43-50.
- Heuser M, Thol F, Ganser A: Ematopoiesi clonale a potenziale indeterminato. Deutsches Arzteblatt international 2016; 113(18): 317-322.
- Fuster JJ, Walsh K: Mutazioni somatiche ed ematopoiesi clonale: nuovi potenziali fattori inaspettati della malattia cardiovascolare legata all’età. Ricerca sulla circolazione 2018; 122(3): 523-532.
- Valent P, et al.: Citopenia idiopatica di significato indeterminato (ICUS) e displasia idiopatica di significato incerto (IDUS) e loro distinzione dalle SMD a basso rischio. Ricerca sulla leucemia 2012; 36(1): 1-5.
- Malcovati L, et al.: Significato clinico della mutazione somatica nella citopenia ematica inspiegabile. Sangue 2017; 129(25): 3371-3378.
- Valent P, et al: Proposta di criteri diagnostici minimi per le sindromi mielodisplastiche (MDS) e potenziali condizioni pre-MDS. Oncotarget 2017; 8(43): 73483-73500.
- Gañán-Gómez I, et al.: Deregolazione della segnalazione immunitaria e infiammatoria innata nelle sindromi mielodisplastiche. Leucemia 2015; 29(7): 1458-1469.
- Glenthøj A, et al: Meccanismi immunitari nella sindrome mielodisplastica. Rivista internazionale di scienze molecolari 2016; 17(6): 944.
- Chung SS, Park CY: Invecchiamento, emopoiesi e sindromi mielodisplastiche. Blood advances 2017; 1(26): 2572-2578.
- Cooper JN, Young NS: Clonalità nel contesto: cloni ematopoietici nel loro ambiente midollare. Sangue 2017; 130(22): 2363-2372.
- Malcovati L, et al: La mutazione SF3B1 identifica un sottogruppo distinto di sindrome mielodisplastica con sideroblasti ad anello. Sangue 2015; 126(2): 233-241.
- Gruppo di studio nordico sulle MDS: Linee guida per la gestione del paziente nelle sindromi mielodisplastiche e nella leucemia mielomonocitica cronica. 8° aggiornamento. www.nmds.org/index.php/guidelines (citato il 14.03.2018).
- Montalban-Bravo G, Garcia-Manero G: Sindromi mielodisplastiche: aggiornamento 2018 su diagnosi, stratificazione del rischio e gestione. Am J Hematol 2018 Jan; 93(1): 129-147.
Ulteriori letture:
- List A, Ebert BL, Fenaux P: Un decennio di progressi nella sindrome mielodisplastica con delezione del cromosoma 5q. Leucemia 2018. DOI: 10.1038/s41375-018-0029-9 [Epub ahead of print].
- Platzbecker U, et al: Luspatercept per il trattamento dell’anemia nei pazienti con sindromi mielodisplastiche a basso rischio (PACE-MDS): uno studio multicentrico, in aperto, di fase 2 di ricerca della dose con studio di estensione a lungo termine. The Lancet Oncology 2017; 18(10): 1338-1347.
InFo ONCOLOGIA & EMATOLOGIA 2018; 6(2): 22-26.