La distruzione cronica dei globuli rossi è caratteristica dell’emoglobinuria parossistica notturna. Le mutazioni nel gene PIG-A portano a un deficit di GPI-AP, che si traduce in un’attivazione incontrollata del complemento. L’inibizione del complemento C5, che deve essere somministrata solo ogni otto settimane per la prima volta, può aiutare.
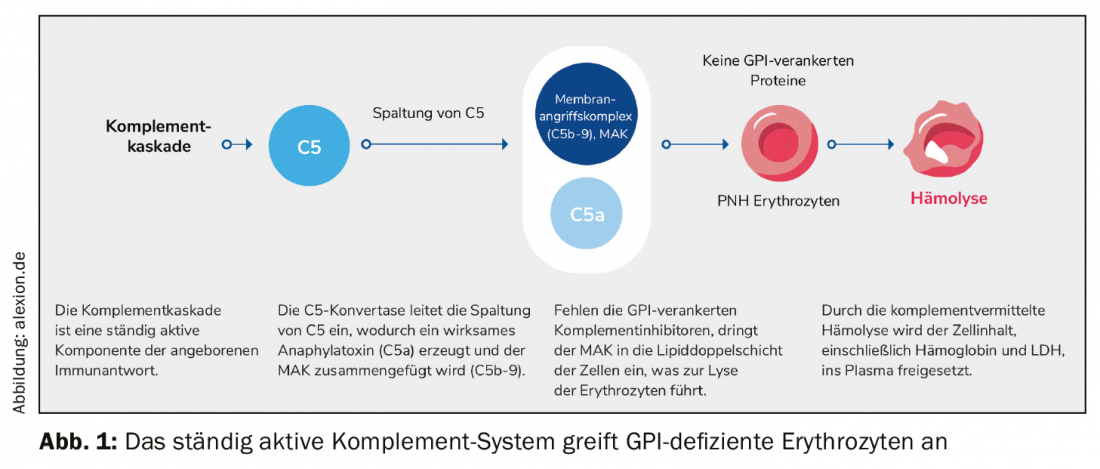
La caratteristica principale della rara emoglobinuria parossistica notturna cronica e progressiva (PNH) è l’emolisi mediata dal complemento. A causa di una mutazione acquisita, le proteine di ancoraggio del glicofosfatidilostiolo (GPI) mancano nelle cellule staminali ematopoietiche del midollo osseo. Di conseguenza, le proteine ancorate al GPI non possono essere formate, soprattutto sulla superficie degli eritrociti rossi. La protezione contro il sistema del complemento, una parte del sistema immunitario dell’organismo, viene meno. I globuli rossi vengono scambiati per invasori, attaccati e distrutti. Il complesso di attacco alla membrana (MAK) penetra nel bilayer lipidico della cellula e innesca la lisi degli eritrociti. L’intero contenuto cellulare, compresa l’emoglobina e la LDH, viene rilasciato nel plasma (Fig. 1).
Malattia complessa con sintomi variabili
La malattia è caratterizzata da anemia emolitica, emoglobinuria e insufficienza o fallimento del midollo osseo, tra gli altri sintomi. Classicamente si manifesta soprattutto con affaticamento, una qualità di vita ridotta e dispnea da sforzo. Tuttavia, possono verificarsi anche i sintomi non specifici che spesso accompagnano le crisi emolitiche. Tuttavia, la causa principale dell’aumento della morbilità e della mortalità dei pazienti è la trombofilia. Fino al 50% di tutte le persone colpite sviluppano trombosi senza misure terapeutiche specifiche. Il 35% dei pazienti muore entro 5 anni dalla diagnosi.
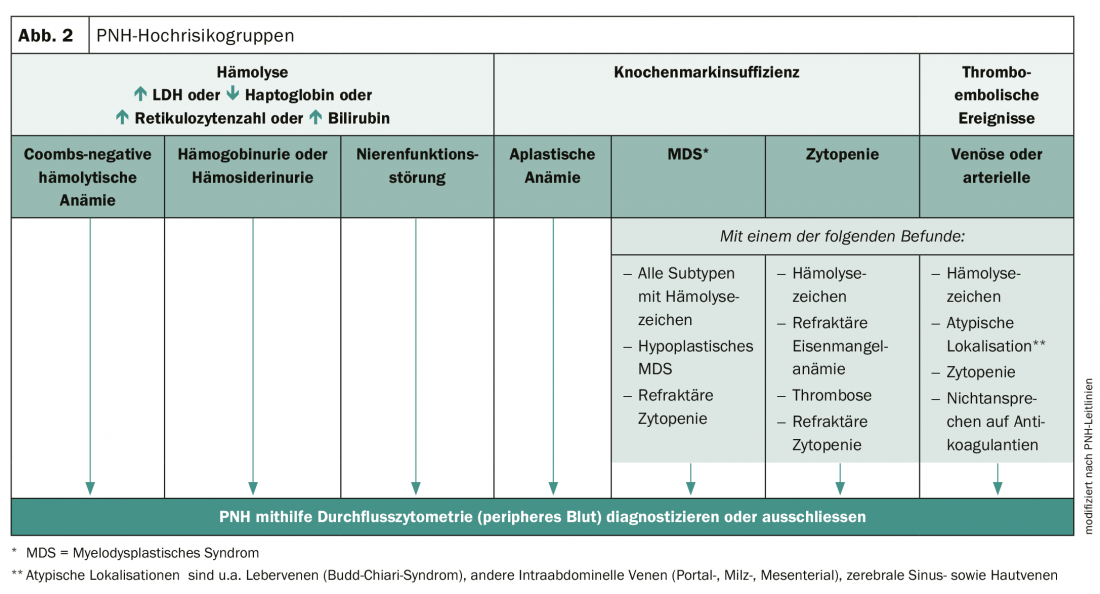
La diagnosi precoce è essenziale per migliorare la prognosi delle persone colpite. La PNH può essere diagnosticata utilizzando la citometria a flusso ad alta sensibilità e un esame clinico completo. Tuttavia, molti medici non sono consapevoli della complessità dei sintomi e della variabilità della malattia. Di conseguenza, la PNH spesso non viene riconosciuta e la diagnosi viene fatta solo dopo un ritardo da uno a più di 5 anni. Tuttavia, ci sono gruppi ad alto rischio in cui la presenza di PNH deve essere considerata (Fig. 2).
La gestione della terapia migliora
Per fermare l’emolisi, le funzioni terminali della cascata del complemento vengono interferite con gli inibitori del complemento. Le funzioni prossimali, invece, rimangono. Per esempio, il tasso di eventi tromboembolici (TE) è stato significativamente ridotto da 11,54 a 0,72 nei pazienti trattati con anticoagulanti. L’inibitore è stato convincente anche in termini di beneficio clinico a lungo termine. L’unico inconveniente: il breve intervallo di infusione. Ora l’inibitore potrebbe essere ulteriormente sviluppato e l’emivita estesa (Ravulizumab). Riducendo il numero di infusioni a 6-7 all’anno, la qualità di vita delle persone colpite è migliorata in modo significativo e anche il rischio di emolisi da rottura è stato ridotto dal 10,7% al 4%.
Fonte: Riunione annuale 2019 delle Società di Ematologia e Oncologia Medica di lingua tedesca (DGHO)
InFo ONCOLOGIA & EMATOLOGIA 2019; 7(6): 32-33 (pubblicato il 6.12.19, prima della stampa).