La sindrome di Lynch, nota anche come HNPCC (cancro colorettale ereditario non poliposico), è la causa ereditaria più comune di cancro colorettale in tutto il mondo. Oltre alla chirurgia preventiva, l’aspirina viene utilizzata anche per la profilassi del cancro. Quando si deve pensare alla sindrome di Lynch e cosa si deve fare se si sospetta?
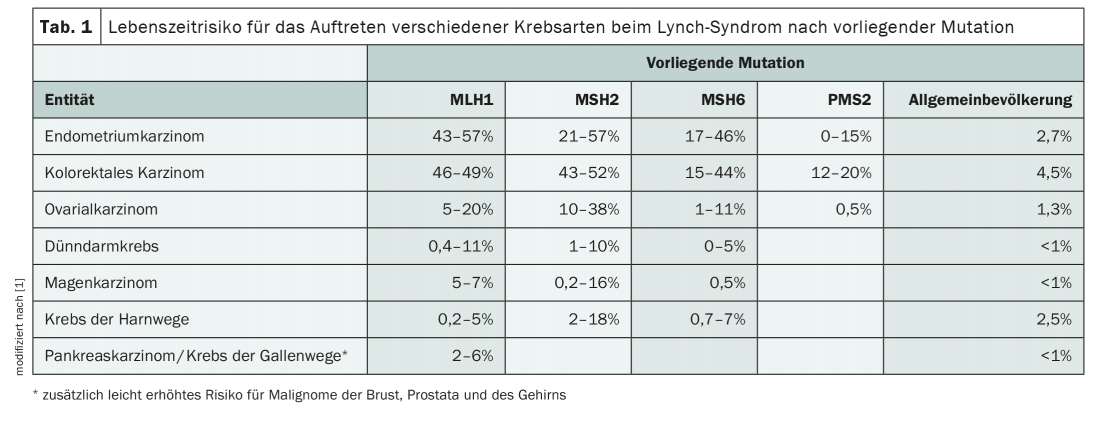
La sindrome di Lynch, che viene ereditata in modo autosomico dominante, aumenta notevolmente il rischio di sviluppare vari tipi di cancro. Oltre ai carcinomi dell’endometrio e del colon-retto, sono frequenti anche altre patologie maligne come i tumori ovarici, pancreatici o gastrici (Tab. 1) . Sebbene le mutazioni che compongono la sindrome HNPCC siano rare nella popolazione, con una prevalenza compresa tra 1:270 e 1:440, rappresentano comunque la predisposizione ereditaria al cancro più frequente in assoluto [1]. La diagnosi precoce e le misure preventive che coinvolgono l’intera famiglia possono prevenire le malattie maligne.
Riparazione difettosa del DNA
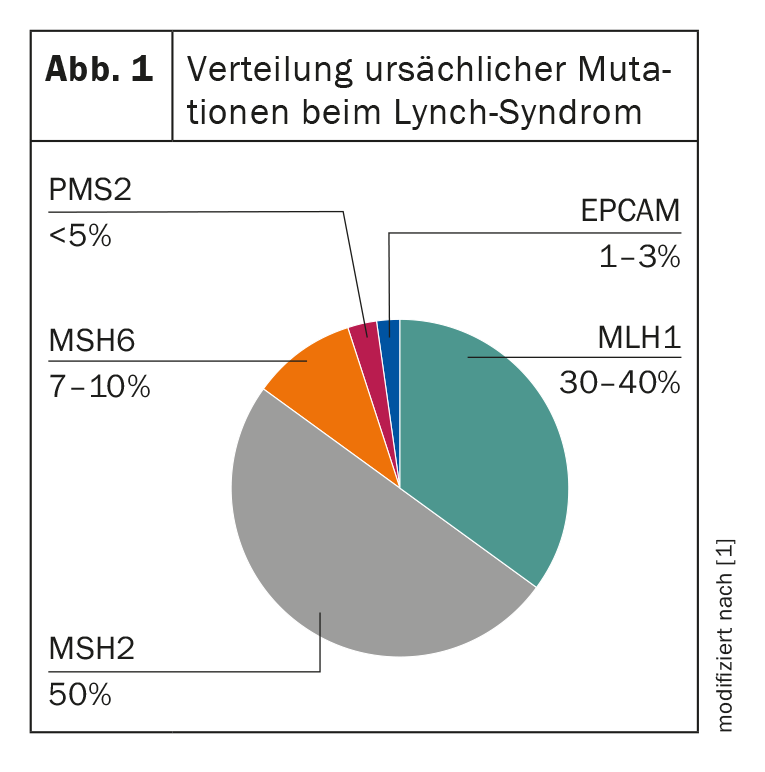
Negli individui affetti, si può rilevare un difetto genetico nella riparazione del DNA, che si riflette in un allungamento dei segmenti di DNA ripetitivi – i cosiddetti microsatelliti. Questi differiscono tra il tessuto tumorale e il tessuto sano nei pazienti con HNPCC, il che si chiama “instabilità microsatellitare” (MSI). La presenza di un MSI di questo tipo, con un sospetto clinico simultaneo, è quindi molto probabile che indichi la sindrome di Lynch [2]. Finora si conoscono quattro geni di riparazione dei mismatch del DNA (MMR) le cui mutazioni germinali portano allo sviluppo dell’HNPCC: MLH1, MSH2, MSH6 e PMS2. Inoltre, una delezione germinale del gene EPCAM è una possibile causa, che comporta anche una perdita della proteina MSH2 nel tumore (Fig. 1) . Una corrispondente perdita di espressione nelle cellule tumorali può essere evidenziata dall’immunoistochimica e la diagnosi viene successivamente confermata dalla genetica molecolare [2]. L’identificazione della mutazione familiare è di grande importanza, poiché non riguarda solo il paziente stesso, ma anche tutta la sua famiglia. Consente di eseguire test predittivi su parenti sani, il che rende possibili le misure preventive in primo luogo. Il rischio di contrarre la malattia varia a seconda della mutazione e dell’età. Questo può essere calcolato individualmente utilizzando il database prospettico della sindrome di Lynch [3].
A causa dell’eredità autosomica dominante, di solito c’è una copia difettosa del gene scatenante nella linea germinale, che viene trasmessa con una probabilità del 50%. Se si verificano mutazioni somatiche, cioè casuali, nella seconda copia genica – originariamente funzionale – si verifica il difetto di riparazione del DNA descritto e quindi una degenerazione maligna accelerata. Quindi, sono necessari ulteriori eventi sfavorevoli perché si sviluppi il cancro. Questo meccanismo spiega l’accumulo di carcinomi del colon-retto in particolare, in quanto la dinamica della formazione dell’adenoma rappresenta probabilmente un fattore di rischio indipendente [4].
Il lupo travestito da pecora
Ma quando ha senso fare un test genetico? Quali sono i segni clinici che suggeriscono la presenza della sindrome di Lynch? A differenza di altre sindromi, come la poliposi adenomatosa familiare (FAP), nella sindrome HNPCC non ci sono chiare caratteristiche fenotipiche che suggeriscano una malattia ereditaria. Di solito ci sono solo singoli adenomi o carcinomi, che sono clinicamente indistinguibili dai tumori sporadici. Solo con la comparsa di diversi tumori maligni, a causa dell’età spesso giovane delle persone colpite o di una storia familiare evidente, si può sospettare la sindrome di Lynch [5]. Ad esempio, l’età mediana della diagnosi di cancro del colon-retto è di 45 anni e in circa il 30% dei casi si aggiunge un altro tumore tipico entro dieci anni [6]. In generale, i tumori associati a HNPCC sono per lo più adenocarcinomi; nell’intestino, si verificano preferibilmente nell’emicolon destro.
Per facilitare l’identificazione dei pazienti a rischio, nel corso degli anni sono stati sviluppati diversi punteggi diagnostici, in particolare i Criteri di Amsterdam e le Linee guida di Bethesda. Inoltre, esiste il modello PREMM per il calcolo delle probabilità. L’elemento centrale di tutti questi sistemi è una storia familiare dettagliata. Per soddisfare i criteri di Amsterdam II, che sono sufficienti per la diagnosi di HNPCC, almeno tre parenti devono avere un cancro associato alla sindrome di Lynch, almeno due generazioni consecutive devono essere colpite e almeno un membro della famiglia deve aver avuto la malattia prima dei 50 anni (panoramica 1) [7]. Nei casi meno chiari, le Linee guida di Bethesda aiutano a identificare le persone potenzialmente colpite e a indirizzarle verso la diagnostica appropriata. (Panoramica 2). Con la crescente disponibilità dell’analisi MSI, che oggi viene già eseguita di routine per molti carcinomi, queste linee guida stanno perdendo la loro importanza, ma sono ancora importanti nella valutazione clinica dei risultati [7]. Questo perché l’instabilità microsatellitare non dimostra la sindrome di Lynch, in quanto si verifica nel 10-15% di tutti i carcinomi del colon e nel 15-20% di tutti i carcinomi endometriali [5].

Instabilità microsatellitare rilevata… e ora?
Se viene rilevata un’instabilità microsatellitare o una carenza delle proteine MMR nel tumore di un paziente che si sospetta abbia la sindrome di Lynch, c’è l’indicazione di eseguire un test genetico per le mutazioni scatenanti. Questi, così come la consulenza genetica, sono servizi obbligatori forniti dalla cassa malattia, a condizione che siano soddisfatte le condizioni per il chiarimento genetico [1]. Se viene trovata una mutazione per la quale è stato stabilito un chiaro legame con la malattia, si consiglia di eseguire un test mirato sui membri della famiglia. In questo contesto, i cambiamenti genetici di importanza (ancora) sconosciuta sono problematici. In questo caso, la prevenzione si basa sulla storia medica personale e sulla storia familiare e non sul risultato del test genetico. La procedura è quindi la stessa che nel caso di un reperto non appariscente; i membri della famiglia non devono essere sottoposti al test.
In assenza di terapie specifiche, il trattamento dei tumori maligni si basa principalmente sulle raccomandazioni che si applicano anche alle malattie tumorali sporadiche. Negli stadi avanzati, tuttavia, gli inibitori del checkpoint sono sempre più utilizzati. Ad esempio, pembrolizumab è approvato come monoterapia per il trattamento di prima linea del tumore del colon-retto metastatico con MSI elevato o riparazione difettosa del DNA mismatch [8]. Per quanto riguarda l’uso di ulteriori immunoterapici e il valore dell’instabilità dei microsatelliti come marcatore predittivo, si possono prevedere dati interessanti nei prossimi anni.
Prevenzione e diagnosi precoce
L’evidenza genetica della sindrome di Lynch si traduce in una lunga coda di topo di raccomandazioni per un ulteriore screening del cancro. Questi si applicano anche ai parenti che sono portatori della mutazione corrispondente. Oltre a uno stile di vita sano e alle misure di diagnosi precoce, si ricorre anche alla chirurgia preventiva e ai farmaci. Colonscopie ed esami ginecologici regolari dovrebbero prevenire il cancro del colon-retto e dell’endometrio. Si raccomanda anche lo screening per l’Helicobacter pylori (Panoramica 3). Attualmente non esiste una diagnosi precoce efficace per il carcinoma ovarico, che si verifica anche frequentemente, o per i tumori uroteliali. Ciò rende ancora più importante la sensibilizzazione delle persone colpite ai sintomi corrispondenti e, a seconda della malattia tumorale della famiglia, l’attuazione di misure aggiuntive per il monitoraggio.
In alcuni casi, l’isterectomia profilattica con o senza annessiectomia viene eseguita dopo aver completato la pianificazione familiare. A volte si ricorre anche a resezioni estese del colon. La conclusione è che il beneficio della chirurgia preventiva per la sindrome di Lynch non è chiaro e deve essere valutato su base individuale [1]. Mancano anche dati affidabili sull’uso profilattico dell’aspirina, soprattutto per quanto riguarda il dosaggio e la durata della somministrazione. Secondo i risultati a lungo termine dello studio CAPP II, l’assunzione giornaliera di 600 mg di acido acetilsalicilico riduce significativamente il rischio di tutti i tumori maligni associati a HNPCC, dopo un periodo di latenza di circa quattro anni. Questo effetto sembra durare per circa dieci anni dopo due anni di somministrazione, anche senza ulteriori somministrazioni [9]. Con CAPP III, è attualmente in corso uno studio per valutare dosi inferiori.
Sebbene negli ultimi anni si sia fatto molto per la diagnosi genetica e l’ulteriore gestione dei pazienti con sindrome di Lynch, soprattutto nei centri, è probabile che la sindrome sia ancora sottodiagnosticata [10]. Per contribuire in modo significativo a una migliore assistenza alle persone colpite, possiamo fare soprattutto una cosa: Ci pensi.
Letteratura:
- SAKK: Guida alla consulenza sulla sindrome di Lynch 2021. www.sakk.ch/sites/default/files/2020-11/Leitfaden%20Lynch-Syndrom.pdf. (ultimo accesso 08.05.2021)
- UKB, Istituto di Genetica Umana: HNPCC / Sindrome di Lynch. www.humangenetics.uni-bonn.de/de/beratung/erbliche-tumorerkrankungen/krankheitsbilder/hnpcc-lynch-syndrom (ultimo accesso 08.05.2021)
- Prospective Lynch Syndrome Database (PLSD) – rischio cumulativo di cancro per età, variante genetica e sesso nei portatori sottoposti a colonscopia. www.plsd.eu.
- Engel C, et al.: Efficacia della sorveglianza colonscopica annuale nei soggetti con cancro colorettale ereditario non poliposico. Clin Gastroenterol Hepatol. 2010; 8(2): 174-182.
- Steinke V, et al: Cancro colorettale ereditario senza poliposi. Dtsch Arztebl International. 2013; 110(3): 32-8.
- Lynch HT, et al: Revisione della sindrome di Lynch: storia, genetica molecolare, screening, diagnosi differenziale e ramificazioni medicolegali. Genetica clinica. 2009; 76(1): 1-18.
- Livstone EM: Sindrome di Lynch 2019. Manuale MSD. www.msdmanuals.com/de/profi/gastrointestinale-erkrankungen/tumoren-des-gastrointestinaltrakts/lynch-syndrom (ultimo accesso 08.05.2021)
- Informazioni sui medicinali di swissmedic, l’Istituto svizzero per gli agenti terapeutici. www.swissmedicinfo.ch (ultimo accesso 08.05.2021)
- Burn J, et al: Prevenzione del cancro con l’aspirina nel cancro colorettale ereditario (sindrome di Lynch), 10 anni di follow-up e dati ventennali basati sul registro nello studio CAPP2: uno studio in doppio cieco, randomizzato, controllato con placebo. The Lancet. 2020; 395(10240): 1855-1863.
- Morrow A, et al: Comprendere il successo dell’implementazione: protocollo per una valutazione approfondita, a metodi misti, del processo di uno studio randomizzato controllato a grappolo che ha testato i metodi per migliorare la rilevazione della sindrome di Lynch negli ospedali australiani. BMJ Open. 2020; 10(6): e033552.
- Vasen HF, et al: Nuovi criteri clinici per il cancro colorettale ereditario non poliposico (HNPCC, sindrome di Lynch) proposti dal gruppo collaborativo internazionale sull’HNPCC. Gastroenterologia. 1999; 116(6): 1453-1456.
- Umar A, et al: Linee guida Bethesda riviste per il cancro colorettale ereditario non poliposico (sindrome di Lynch) e l’instabilità microsatellitare. J Natl Cancer Inst. 2004; 96(4): 261-268.
InFo ONCOLOGIA & EMATOLOGIA 2021; 9(3): 37-39