Il termine linfomi indolenti copre un numero crescente di linfomi non Hodgkin a bassa malignità e leucemie, prevalentemente della serie B. Vengono differenziati e classificati l’uno dall’altro in base alla costellazione di marcatori e al profilo genetico. Il trattamento dipende dall’estensione o dalla dinamica della malattia e dai sintomi del paziente. Attualmente, la terapia si sta spostando dai classici chemioterapici agli inibitori del segnale e agli immunoterapici. L’obiettivo primario del trattamento è spesso quello di controllare la malattia il più a lungo possibile, con una tolleranza accettabile della terapia.
Il termine linfoma non-Hodgkin indolente (iNHL) copre un gruppo biologicamente eterogeneo di linfomi maturi e a piccole cellule della serie B con una tendenza alla manifestazione leucemica. In passato, il termine “indolente” descriveva un gruppo di linfomi a crescita lenta, “poco maligni”, i cui sottotipi erano spesso difficili da separare l’uno dall’altro e che non sempre dovevano essere differenziati l’uno dall’altro a causa della mancanza di opzioni terapeutiche.

Nel frattempo, i dati immunofenotipici e molecolari ampliati consentono una migliore suddivisione in entità di linfoma, che dovrebbe essere effettuata secondo l’attuale classificazione dei linfomi dell’OMS 2008 (Tab. 1) [1]. Ci sono iNHL che nel corso della malattia si trasformano in linfomi blastici o aggressivi altamente maligni, primo fra tutti il linfoma follicolare, seguito dalla leucemia linfocitica cronica (LLC). Inoltre, esistono entità come il linfoma a cellule del mantello (MCL), la maggior parte delle quali (circa il 90% di tutti i casi) presenta un comportamento di crescita aggressivo e viene trattata con un’immuno-chemioterapia intensiva (se necessario con una terapia ad alte dosi e la sostituzione di cellule staminali autologhe). Ma c’è anche un sottogruppo indolente che si manifesta soprattutto nei pazienti anziani, con un coinvolgimento del midollo osseo e della milza. Senza dimenticare i tipici NHL che possono presentarsi in un contesto clinico specifico, come il MALT associato all’Helicobacter pylori o il linfoma della zona marginale associato all’HCV.
Diagnosi e sottotipi
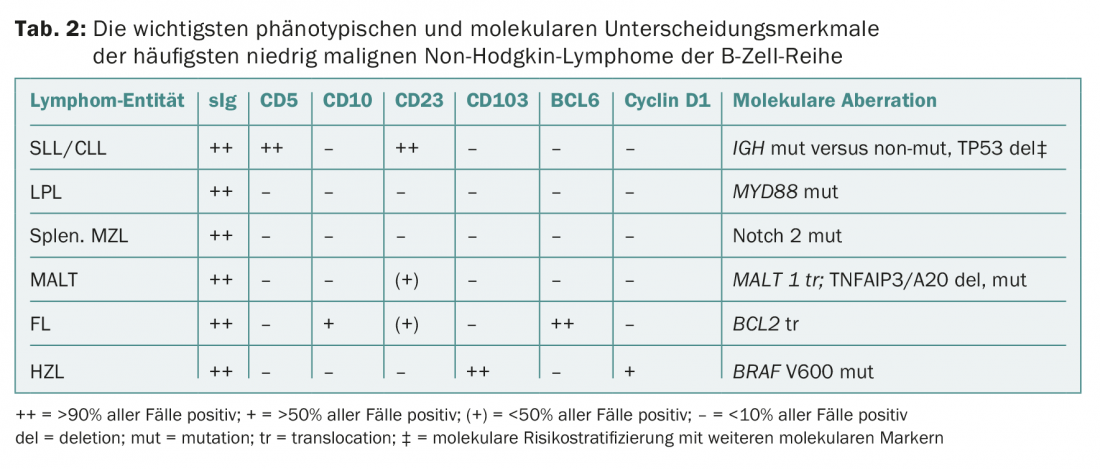
La diagnosi viene effettuata mediante biopsia dei linfonodi e/o del midollo osseo, fenotipizzazione (immunitaria) del sangue e/o del midollo osseo e analisi genetico-molecolari (Tab. 2) . Questi test sono standardizzati e si applicano a tutte le patologie di linfoma. Secondo la classificazione dell’OMS, le seguenti entità appartengono all’iNHL:
- Leucemia linfatica cronica (LLC) resp. la sua forma nodale (linfoma linfocitico a piccole cellule, SLL)
- Linfoplasmacitico (LPL o malattia di Waldenström)
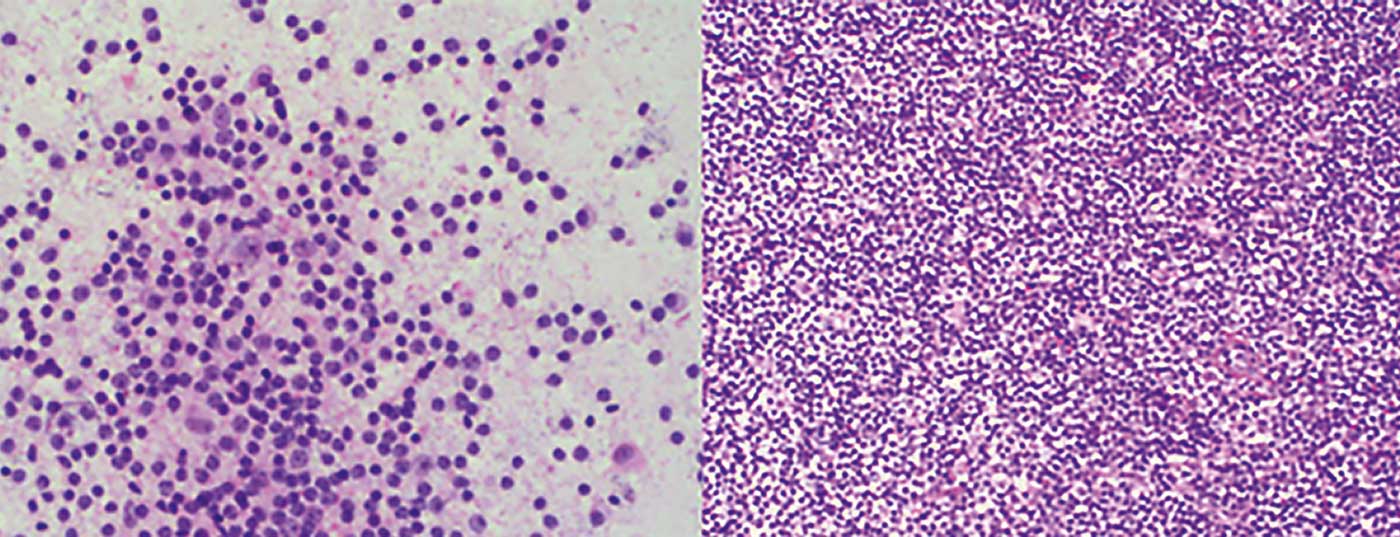
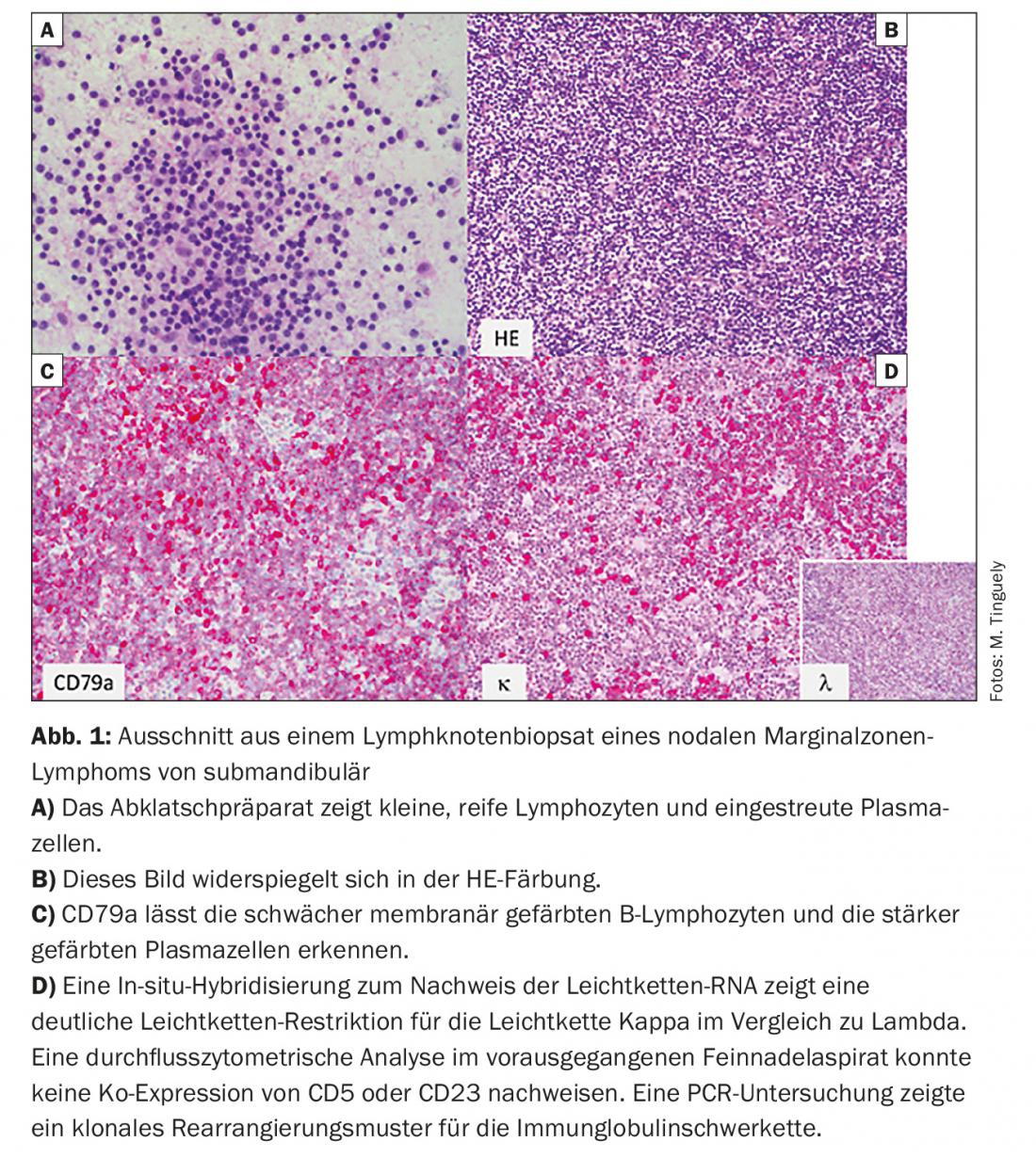
- Il linfoma della zona marginale (MZL) (Fig. 1) con i suoi tre sottotipi: nodale, extranodale (ad esempio MALT) e splenico.
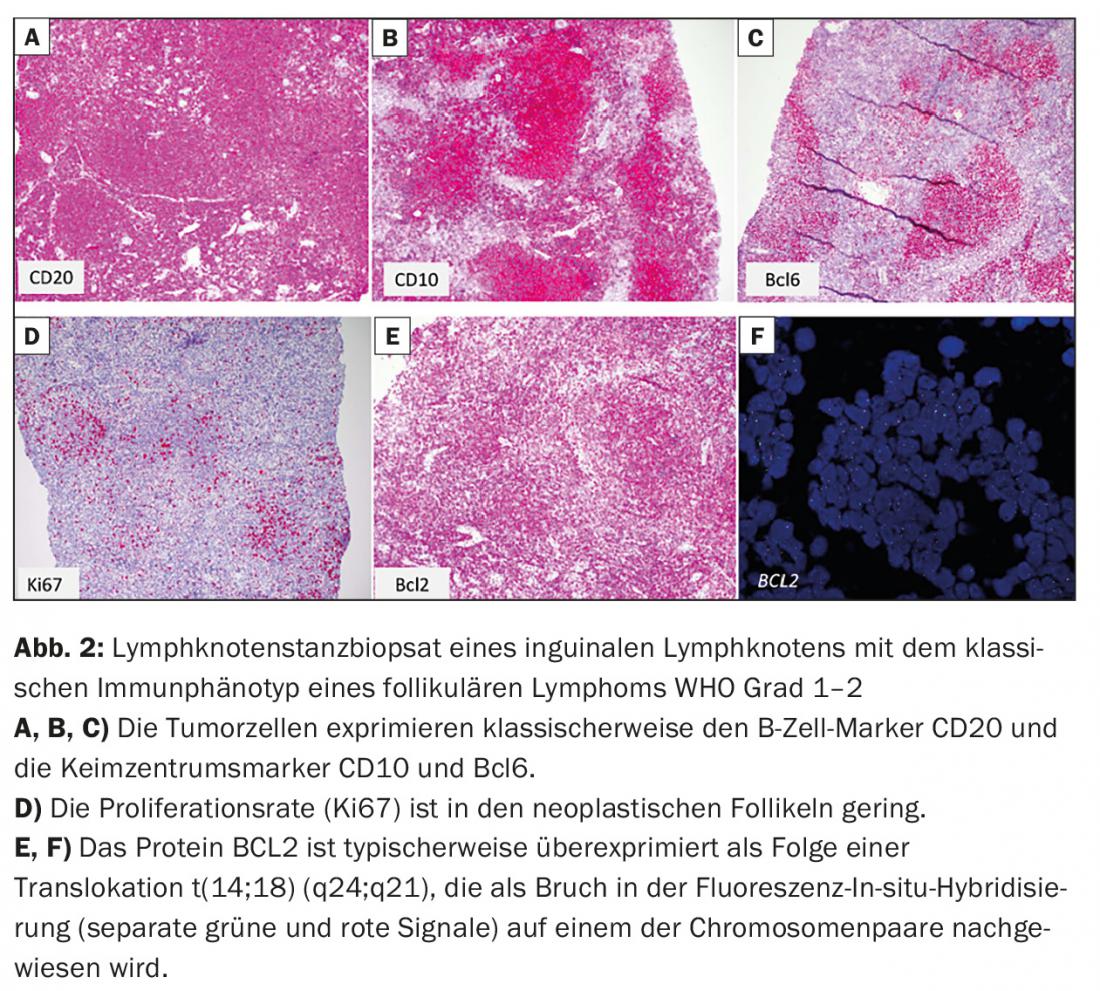
- Linfoma follicolare (FL) (Fig. 2)
- Leucemia a cellule capellute (HZL).
La LLC e la leucemia a cellule capellute non sono considerate da tutti i gruppi di lavoro come facenti parte dell’iNHL perché, come suggerisce il nome, spesso si manifestano principalmente con alterazioni dell’emocromo e in alcuni casi (questo vale in particolare per la leucemia a cellule capellute) richiedono altre misure terapeutiche. Il linfoma a cellule del mantello (MCL) non dovrebbe più essere conteggiato come iNHL per le ragioni menzionate, sebbene esista un sottogruppo indolente nei pazienti più anziani.
Presentazione clinica e principi terapeutici
I linfomi indolenti sono di solito una malattia dell’età avanzata. La stragrande maggioranza dei pazienti (66%) si trova già in stadi avanzati III e IV (stadiazione secondo la classificazione di Ann Arbor o, più recentemente, di Lugano) al momento della diagnosi e quindi non è più suscettibile di terapia curativa [2]. La diagnosi non è sempre associata alla necessità di iniziare immediatamente la terapia. Anche se le raccomandazioni variano un po’ in base ai singoli sottotipi, è comunque vero che si deve trattare solo la malattia sintomatica. La definizione di malattia sintomatica è rimasta in gran parte invariata negli ultimi 30 anni e comprende i sintomi locali dovuti alla crescita del linfoma, la compromissione della normale funzione degli organi (ad esempio, l’anemia sintomatica), i sintomi B, la malattia extranodale sintomatica, altre citopenie o un rapido tasso di crescita di una manifestazione di linfoma. Questi criteri sono molto elastici e offrono al paziente e al medico un ampio margine decisionale. Pertanto, sono in corso sforzi per stabilire una classificazione prognostica migliore e quindi un supporto decisionale attraverso nuovi marcatori (molecolari). Un esempio è l’indice prognostico della CLL (CLL-IPI), presentato di recente, che fornisce una raccomandazione sulla tempistica e sul tipo di terapia [3].
Radioterapia nelle fasi iniziali: un’opzione curativa?
La radioterapia è stata definita negli ultimi decenni come un’opzione di trattamento curativo per gli stadi iniziali del linfoma follicolare. Il 15-25% di tutti i pazienti con diagnosi di linfoma follicolare presenta uno stadio I o II e le attuali linee guida nazionali e internazionali raccomandano ancora la radioterapia per questi pazienti [4].
La difficoltà di una raccomandazione positiva per l’implementazione della radioterapia nella routine clinica odierna si basa sull’incertezza se i dati dei vecchi studi (superfici di irradiazione di grandi dimensioni, dosi totali elevate, applicazioni non specifiche ai linfonodi o ai campi involuti) possano essere trasferiti alle tecniche di irradiazione odierne. Questo pone il dilemma principale: attualmente non siamo in grado di definire il presunto valore curativo della radioterapia per i linfomi indolenti (principalmente il linfoma follicolare) nelle fasi iniziali.
Attesa dopo la diagnosi: è ancora valida?
Alcuni autori ritengono che attualmente circa il 50% dei pazienti non necessiti di un trattamento immediato al momento della diagnosi. Il termine “watch & wait” (w&w) è stato coniato più di 35 anni fa [5]. L’attesa sembrava giustificabile quando la malattia progrediva lentamente e non c’erano sintomi o c’erano solo sintomi minori – e anche perché non esistevano terapie efficaci. A quel tempo, il tempo mediano alla prima terapia per i pazienti con FL era di 31-36 mesi. Le analisi osservazionali longitudinali hanno mostrato che circa il 20% dei pazienti con FL non ha richiesto la terapia a un follow-up mediano di 17 anni. Rispetto a una coorte di pazienti trattati con chemioterapia alla diagnosi, non c’è stata alcuna differenza nella sopravvivenza globale (OS) a 5 anni. La OS mediana era di undici anni e variava molto tra le istologie.
Se il w&w è appropriato riguarda tutti gli iNHL, ma viene analizzato principalmente a livello di FL. Così, in uno studio multicentrico, 379 pazienti sono stati randomizzati in tre bracci di trattamento: sola osservazione (w&w), sola induzione di rituximab (applicazioni quadrisettimanali) o induzione di rituximab seguita da due anni di terapia di mantenimento (rituximab ogni due mesi) [6]. Questo ha mostrato un vantaggio significativo nella sopravvivenza libera da progressione (PFS) per entrambi i bracci di rituximab rispetto al w&w. Tuttavia, la sopravvivenza complessiva non differiva, per cui ancora oggi non esiste una ragione convincente per l’uso precoce dell’immunoterapia. Questo potrebbe cambiare con i nuovi farmaci (immunoterapici) e quindi dovrebbe essere sempre messo in discussione.
Terapie attuali
Le opzioni di trattamento per l’iNHL sono cambiate radicalmente negli ultimi anni con l’introduzione di nuove terapie. Questi si basano principalmente sugli anticorpi e sull’inibizione della tirosin-chinasi (Fig. 3). Finora, l’immunoterapia con anticorpi specifici per il CD20 come monoterapia o in combinazione con agenti chemioterapici classici ha dominato la terapia di prima linea. Si deve ancora presumere che una cura non sia possibile, nonostante i moderni metodi terapeutici; l’unica eccezione è il trapianto di cellule staminali allogeniche.
Anticorpi monoclonali specifici per CD20
Un esempio classico di anticorpo specifico per il CD20 è il rituximab, che oggi è parte integrante del trattamento del linfoma a cellule B e può essere utilizzato per diverse entità sia come monoterapia (ad esempio, FL) che in combinazione con la chemioterapia (ad esempio, CLL). Gli anticorpi anti-CD20 più recenti (ad esempio, obinutuzumab) hanno una tossicità cellulare diretta ancora più elevata (attività ADCC) e possono quindi eliminare le cellule del linfoma in modo più efficiente [7]. Nel trattamento della LLC, ad esempio, questo comporta un aumento dei pazienti con malattia minima residua (MRD) negativa. Ciò significa che nei pazienti con obinutuzumab + chemioterapia, la malattia – rispetto al trattamento convenzionale con rituximab + chemioterapia – è più spesso non rilevabile (dal 38 al 3% nel sangue periferico), nonostante i metodi di rilevamento sensibili. Resta da vedere se l’aumento del tasso di negatività della MRD porterà anche a un prolungamento della sopravvivenza.
Combinazione di anticorpi e chemioterapia
In genere, gli anticorpi specifici per il CD20 vengono utilizzati in combinazione con clorambucile, bendamustina o CHOP. Il clorambucile è spesso utilizzato in combinazione con obinutuzumab nel trattamento dei pazienti anziani con LLC o con rituximab nel linfoma MALT [8]. La bendamustina, invece, in combinazione con rituximab, è considerata lo standard di prima linea nel linfoma follicolare (gradi 1 e 2) [9] e nella LLC (con riserve sulla fludarabina-endoxan). Il rituximab con CHOP è usato un po’ meno frequentemente rispetto al passato e viene utilizzato principalmente per le varianti del linfoma blastoide o per il FL di grado 3 . In generale, il numero di cicli di chemioterapia è spesso limitato a sei (ogni tre o quattro settimane) e l’immunoterapia viene somministrata per lo stesso periodo di tempo o come monoterapia per due anni come terapia di mantenimento.
Immunomodulatori
Gli immunomodulatori (IMID) sono piccole molecole che di solito vengono assunte per via orale. Gli IMID, come la lenalidomide, sono stati utilizzati per la prima volta con successo nel mieloma multiplo e ora vengono testati anche nel linfoma, con tassi di risposta incoraggianti. La combinazione di lenalidomide e rituximab nei pazienti con FL di grado 3 e malattia recidivata o refrattaria ha raggiunto un tasso di risposta globale (ORR) fino all’86%. Quando la combinazione viene utilizzata direttamente nella terapia di prima linea, si possono ottenere ORR fino al 98% con alti tassi di remissione completa (CR) (87% CR e CR non confermata) e negatività della MRD. Recentemente è stata concessa l’approvazione anche per il trattamento del linfoma a cellule mantellari (MCL). L’approvazione si basa su studi con malattia MCL recidivata o refrattaria, grazie a un ORR del 42-53% [10,11].
Inibizione della via di segnalazione del recettore delle cellule B (BCR)
Tassi di risposta ancora più elevati nel trattamento dell’MCL possono essere ottenuti con composti che inibiscono la via di segnalazione a valle del BCR, ad esempio gli inibitori della tirosin-chinasi di Bruton (BTK) o della PI3 chinasi (PI3K) [12]. Entrambe le chinasi sono spesso costitutivamente attive nelle cellule di linfoma e promuovono la proliferazione o la sopravvivenza cellulare.
Inibitori di BTK
Ibrutinib è il primo inibitore BTK approvato che si lega in modo irreversibile e covalente a un residuo di cisteina (Cys-481) della tirosina chinasi, provocando un’inibizione forte e prolungata dell’attività enzimatica. Nella LLC, ibrutinib ha mostrato un’elevata attività. L’approvazione si basa su uno studio comparativo con l’anticorpo monoclonale CD20-specifico ofatumumab (studio RESONATE) in 391 pazienti con CLL/SLL pre-trattati [13]. Ad un follow-up mediano di 9,4 mesi, ibrutinib (420 mg/d/po) ha migliorato significativamente la PFS e la OS. Dopo dodici mesi, la OS era del 90% nel gruppo ibrutinib e dell’81% nel gruppo ofatumumab. Il tasso di risposta globale è stato significativamente più alto per ibrutinib (42,6 vs. 4,1%, p <0,001). Il tasso di risposta e la durata erano indipendenti dalla presenza di del17p o dalla resistenza agli analoghi della purina. Questo dimostra l’alto valore degli inibitori BTK in questo sottogruppo di pazienti con LLC difficile da trattare. Gli eventi avversi più comuni sono stati diarrea, affaticamento, febbre e nausea.
La seconda entità approvata in Svizzera riguarda il trattamento dell’MCL recidivato. La base per l’approvazione è stata uno studio multicentrico, a braccio singolo, di fase II su 111 pazienti con MCL pretrattati con un dosaggio di 560 mg di ibrutinib una volta al giorno. La pubblicazione completa riporta un ORR del 66% con un tasso di CR del 17% e una durata mediana della risposta (DOR) di 17,5 mesi [4]. È interessante notare che il tasso di risposta è aumentato continuamente nel corso del trattamento (la cosiddetta “risposta incrementale nel corso del trattamento”), per cui – a differenza delle chemioterapie classiche – le remissioni tardive possono verificarsi anche con la prosecuzione della terapia.
Inibitori PI3K
La famiglia PI3K è composta da una serie di serina/treonina chinasi che regolano la crescita, la differenziazione, il metabolismo, la sopravvivenza e la proliferazione in varie cellule. L’inibizione dell’unità p110δ, per esempio, porta a una significativa deplezione delle cellule B e al blocco della via di segnalazione a valle del BCR. Pertanto, la maggior parte degli approcci terapeutici nel trattamento del linfoma si concentra sul blocco diretto dell’unità p110δ. Il prototipo di questa classe di sostanze è idelalisib, un inibitore selettivo di p110δ.
Idelalisib è stato inizialmente testato in uno studio randomizzato di fase III su 220 pazienti con CLL recidivata in combinazione con rituximab [15]. Nel braccio di controllo, i pazienti hanno ricevuto rituximab più placebo. Con un pretrattamento mediano con tre sostanze, in quasi tutti i casi sono stati utilizzati prima il rituximab e un analogo alchilano o nucleotide purinico. Quasi il 40% dei pazienti con CLL presentava anche un’alterazione genetica sfavorevole del gene p53.
In termini di efficacia, si è registrato un tasso di risposta significativamente più alto nel braccio rituximab + idelalisib (81 vs. 13%), un prolungamento significativo della PFS a 1 anno (93 vs. 46%, p<0,001) e un prolungamento significativo della OS a 1 anno (92 vs. 80%, p=0,02). La superiorità della combinazione di rituximab + idelalisib è stata dimostrata per tutti i sottogruppi.
Gli effetti collaterali che sono significativi per la pratica clinica quotidiana e che vanno oltre l’applicazione del rituximab sono la diarrea precoce e talvolta tardiva. Tuttavia, se si confronta il profilo degli effetti collaterali con altre sostanze che potrebbero essere utilizzate in questa situazione, come ofatumumab, alemtuzumab o farmaci citostatici convenzionali, idelalisib ottiene sicuramente un punteggio positivo.
Come agente monoterapico, idelalisib mostra un’elevata efficacia nell’iNHL nel contesto della terapia delle ricadute. La sostanza è approvata per il trattamento di pazienti affetti da FL con malattia recidivata che hanno ricevuto due precedenti linee di terapia [16]. Nello studio sottostante di fase II a braccio singolo su 125 pazienti affetti da iNHL resistenti al rituximab e agli alchilanti, idelalisib è stato somministrato a 150 mg due volte al giorno fino alla progressione della malattia. Il tempo mediano alla risposta è stato di 1,9 mesi, la durata mediana della risposta è stata di 12,5 mesi e la PFS mediana di 11 mesi. Gli eventi avversi più comuni di grado 3 o superiore sono stati la neutropenia (27% dei pazienti), l’innalzamento delle aminotransferasi (13%), la diarrea (13%) e la polmonite (7%).
Inibitori di BCL-2
La proteina anti-apoptotica BCL-2 è sovraespressa nelle cellule di linfoma e contribuisce alla resistenza alla chemioterapia. Gli inibitori selettivi di BCL-2, somministrati per via orale, come il venetoclax, bloccano la proliferazione delle cellule di linfoma e quindi portano alla remissione del tumore nei modelli preclinici. In uno studio clinico iniziale su 106 pazienti con MCL (n=28), linfoma follicolare (n=29), linfoma diffuso a grandi cellule B (n=41) e altri sottotipi di NHL (n=8), la monoterapia con venetoclax ha mostrato un profilo di sicurezza accettabile con la dose massima tollerata di 1200 mg/d [17]. L’ORR è stato del 44% per tutti i sottotipi, del 78% per l’MCL e del 38% per l’FL. La PFS mediana è stata di 10-14 mesi. Gli eventi avversi più comuni correlati al trattamento (AEs ≥20%) sono stati nausea (48%), diarrea (44%), affaticamento (41%), diminuzione dell’appetito (21%) e vomito (21%). Significativa è la comparsa della sindrome da lisi tumorale, che tuttavia si è verificata in due pazienti senza conseguenze cliniche.
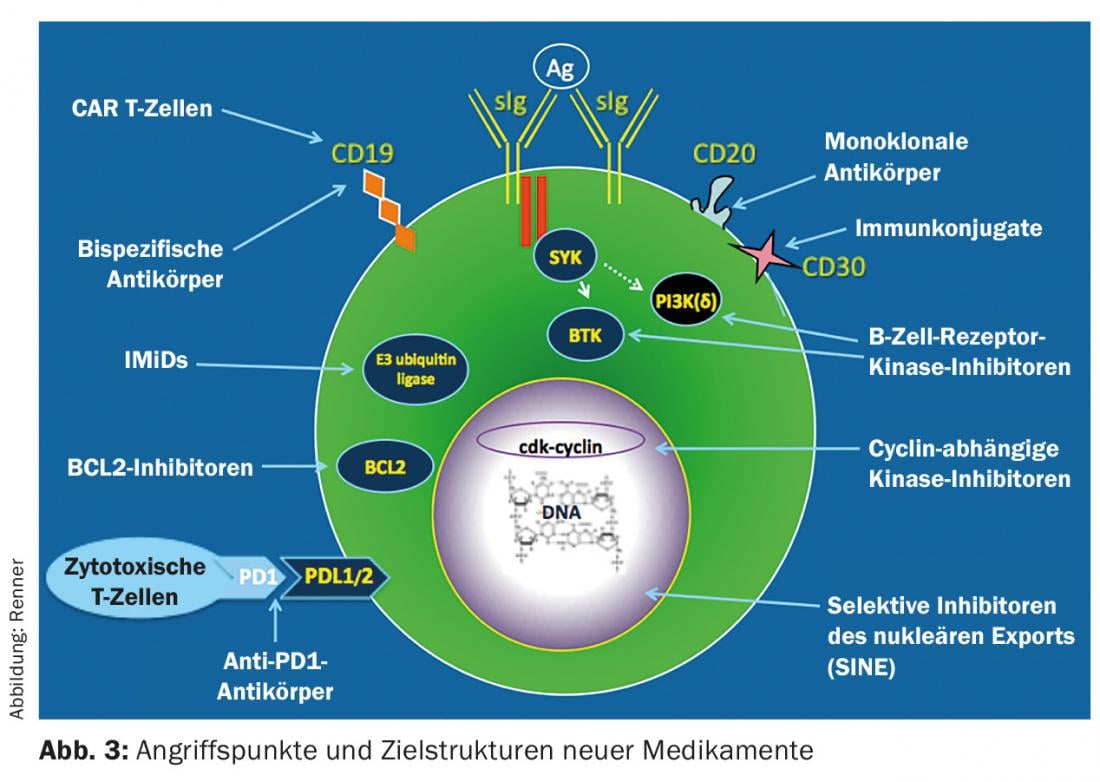
Terapie future
Le terapie future proverranno principalmente da due aree (Fig. 3) : Inibitori della via di segnalazione che mirano a molecole di commutazione importanti della cellula del linfoma e bloccano la loro funzione (ad esempio, gli inibitori CDK 4/6), e nuovi immunoterapici. La consapevolezza che il sistema immunitario contribuisce al controllo del tumore sta attualmente rivoluzionando le opzioni di trattamento emato-oncologico. Gli inibitori del blocco del checkpoint, gli anticorpi bispecifici e le cellule T riprogrammate (cellule T CAR) sono in fase di sviluppo clinico con risultati impressionanti. Forse un giorno riusciremo a stimolare il sistema immunitario in modo tale da rendere possibile il controllo del tumore a lungo termine e persino la guarigione.
Letteratura:
- Swerdlow SH, et al.: IARC Press, Lione, 2008.
- Brice P, et al: J Clin Oncol 1997; 15(3): 1110-1117.
- Molica S, et al: Abstract 498, presentato al 57° Meeting annuale della Società Americana di Ematologia (ASH), 2015.
- Hiddemann W, et al: Internist (Berl) 2016; 57(3): 222-229.
- Morrison VA, Peterson BA: Leuk Lymphoma 1993; 10 Sup: 29-33.
- Adreshna KM, et al: Lancet Oncol 2014; 15(4): 424-435.
- Goede V, et al: NEJM 2014; 370(12): 1101-1110.
- Zucca E, et al: J Clin Oncol 2013; 31(5): 565-572.
- Rummel MJ, et al: Lancet 2013; 381(9873): 1203-1210.
- Habermann TM, et al: Br J Haematol 2009; 145(3): 344-349.
- Witzig TE, et al: Ann Oncol 2011; 22(7): 1622-1627.
- Mato A, et al: Am J Hematol 2015; 90(7): 657-664.
- Byrd JC, et al: NEJM 2014; 371(3): 213-223.
- Wang ML, et al: NEJM 2013; 369(6): 507-516.
- Furman RR, et al: NEJM 2014; 370(11): 997-1007.
- Gopal AK, et al: NEJM 2014; 370(11): 1008-1018.
- Roberts AW, et al: NEJM 2016; 374(4): 311-322.
InFo ONCOLOGIA & EMATOLOGIA 2016; 4(2): 11-15