I depositi di mucina sono possibili in tutti gli strati della pelle, la posizione del deposito non è patognomica. Oltre alle mucinosi primarie, questa dermatosi da deposito si manifesta spesso come epifenomeno. L’eziopatogenesi non è del tutto chiara.
Alla ZDFT di quest’anno, il Prof. Reinhard Dummer, MD, Primario della Clinica Dermatologica dell’Ospedale Universitario di Zurigo, e il suo team (Dr. Thierry Nordmann, MD, Dr. Carole Guillet, MD) hanno parlato delle mucinosi [1] come parte del tema principale delle dermatosi da deposito. La mucina depositata è una sostanza costituita da una miscela di glicosaminoglicani che si trovano anche nella pelle sana in forma libera (acido ialuronico) o legata (proteoglicani). I glicosaminoglicani sono elementi del tessuto connettivo che legano l’acqua e svolgono un ruolo importante nella consistenza e nel turgore della pelle e sono prodotti dai fibroblasti o dai cheratinociti. A livello patogenetico, si verifica un aumento della sintesi o un’alterata degradazione dei glicosaminoglicani.
Sfida differenziale
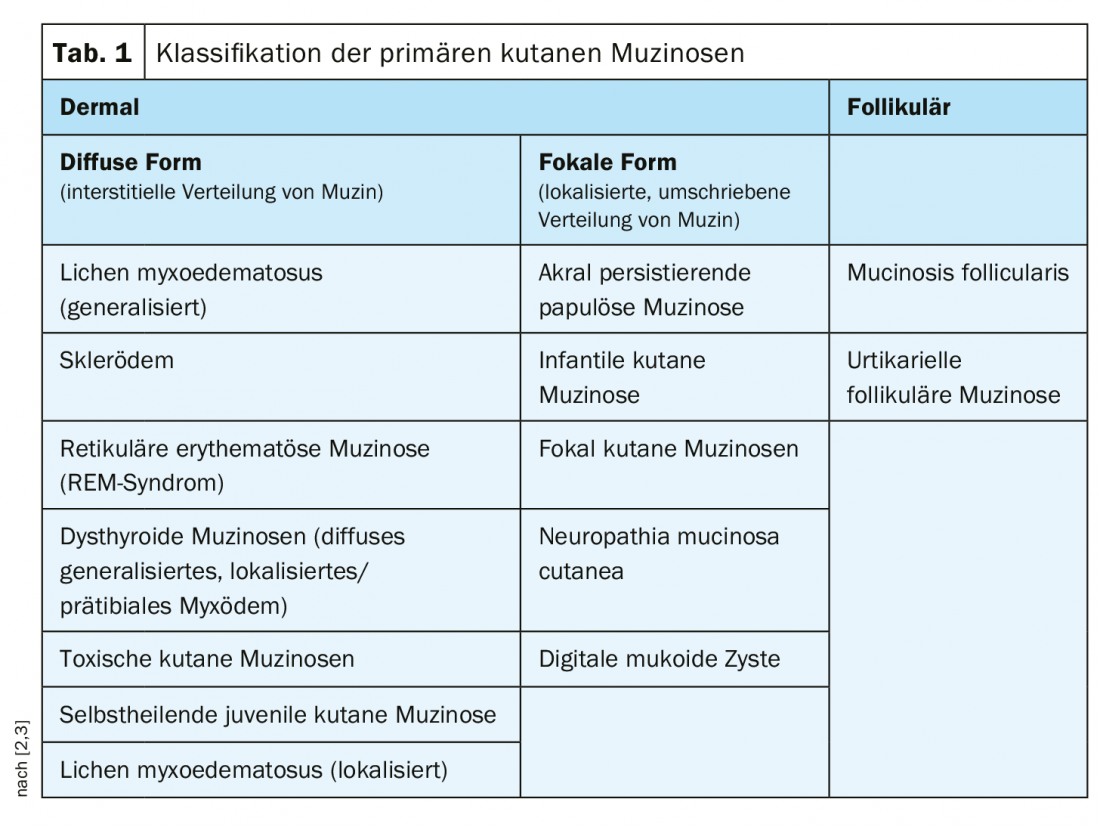
Nelle mucinosi cutanee primarie, si distingue tra sottotipi cutanei e follicolari; i sottotipi cutanei si dividono in una forma diffusa (distribuzione interstiziale della mucina) e una forma focale (distribuzione circoscritta e localizzata della mucina) (Tab. 1) [2,3]. Le mucinosi primarie sono piuttosto rare; più frequente è la comparsa di questa dermatosi da deposito nel contesto di processi infiammatori e proliferativi (fenomeno reattivo). Nel lupus eritematoso e anche nella dermatosi granulomatosa granuloma anulare (DD necrobiosi lipoidica), un aumento della presenza di mucina è un criterio diagnostico differenziale. Ma la proliferazione della mucina può verificarsi anche in molti altri processi infiammatori. Inoltre, nello stroma tumorale dei tumori mesenchimali e annessiali, il tessuto connettivo può essere ricco di mucina. Una classificazione delle mucinosi dermiche come sintomo di varie malattie primarie è riassunta nella tabella 2.

La sindrome REM: controversia sull’eziopatogenesi
La mucinosi eritematosa reticolare, nota anche come sindrome REM, è stata descritta per la prima volta nel 1974 [1,3]. L’eziopatogenesi della sindrome REM non è stata completamente chiarita fino ad oggi; tra l’altro, si discute se possa essere una variante clinica del lupus eritematoso cutaneo [3]. Le caratteristiche cliniche tipiche sono placche striate, reticolari, orticarioidi, leggermente infiltrate e arrossate nell’area centrale superiore del tronco, che nella maggior parte dei casi colpiscono donne giovani [3]. L’esposizione alla luce solare può provocare un aggravamento dei sintomi [1]. In termini di manifestazione clinica, ci sono sovrapposizioni tra la sindrome REM e il lupus eritematoso tumido (forma progressiva di lupus eritematoso cutaneo) e l’infiltrazione linfocitaria di tipo Jessner e Kanof. Finora, non è chiaro fino a che punto queste malattie rappresentino entità indipendenti [3]. I fibroblasti dei pazienti REM mostrano una risposta deviante rispetto alla norma alla stimolazione con IL-1β esogena, che svolge un ruolo nel metabolismo dell’acido ialuronico forse disregolato nella sindrome REM [1]. Le caratteristiche diagnostiche differenziali tra la sindrome REM e il lupus tumidus sono riassunte nella tabella 3.

Lichen myxoedematosus: notare le diagnosi di esclusione
Il lichen myxoedematosus è una rara mucinosi primaria con un decorso cronico progressivo [3]. Si distinguono due sottotipi: la forma localizzata e la forma generalizzata (Tab. 4) [1]. Quest’ultimo è chiamato anche scleromixoedema ed è caratterizzato da caratteristiche sclerodermiformi e papulari.
I siti di predilezione del lichen myxoedematosus localizzato sono il dorso delle mani, i lati estensori delle braccia, la parte superiore del tronco, il viso e le ascelle [3]. Le caratteristiche tipiche sono papule individuali di colore cutaneo, rossastre, che possono confluire e apparire come lichenificazione e/o ispessimento generale della pelle. La distribuzione della mucina nella forma localizzata può essere diffusa o focale. In letteratura c’è una controversia se il lichen myxoedematosus discreto, la mucinosi papulare acrale persistente, la mucinosi papulare autorigenerante (forma giovanile/adulta), la mucinosi infantile e la forma nodulare debbano essere considerati sottotipi distinti di lichen myxoedematosus localizzato. I sintomi classici del sottotipo generalizzato (scleromixoedema) sono l’ispessimento eritematoso generalizzato della pelle, che può portare a mimare la rigidità e la limitazione dei movimenti articolari; in alcuni casi, sono state osservate anomalie cardiovascolari oltre a miopatie e sintomi neurologici [3]. La diagnosi differenziale dello scleromixoedema deve essere differenziata clinicamente e istologicamente da altre alterazioni papulari come la sclerodermia sistemica e lo scleroedema. Una caratteristica che distingue le altre mucinosi sono i reperti istologici caratteristici del lichen myxoedematosus locale e dello scleromixoedema. Queste sono caratterizzate, tra l’altro, da una pronunciata deposizione di mucina nel derma medio e superiore e spesso da un aumento dei fibroblasti con nuclei grandi e a forma di stella; inoltre, le fibre di collagene possono essere compattate e aumentate (fibromucinosi) e può esserci un piccolo infiltrato linfocitario perivascolare [3]. I glucocorticoidi topici e gli inibitori topici della calcineurina sono raccomandati come terapia per il lichen myxoedematosus; possono essere utilizzati anche i trattamenti PUVA e laser. In alcuni casi, la riduzione dei sintomi è stata ottenuta con i citostatici (ciclofosfamide, clorambucile, melfalan), l’isotretinoina e la plasmaferesi, e in alcuni casi con le immunoglobuline per via endovenosa, il melfalan ad alte dosi e il trapianto autologo di cellule staminali del sangue periferico [3]. Il docente sottolinea che in presenza di lichen myxoedematosus localizzato, la malignità e la malattia della tiroide sono diagnosi importanti da escludere [1].

Mucinosi follicolare: ostia follicolare prominente
A differenza della sindrome REM e del lichen myxoedematosus, che sono sottotipi cutanei, la mucinosi follicolare è istologicamente caratterizzata da depositi di mucina nei follicoli. C’è anche un infiltrato linfocitario atipico e una degenerazione dell’epitelio [1]. Clinicamente, il sottotipo follicolare si manifesta con papule e placche focali, follicolari, da color pelle a eritematose, con ostia follicolare prominente (a volte con chiazze cheratotiche simili a comedoni). Dal punto di vista eziopatogenetico, l’infiammazione linfocitaria iniziale viene discussa come possibile causa dell’accumulo secondario di mucina. Si pensa che ci sia una transizione continua dalla mucinosi follicolare (più comune nei pazienti giovani) alla micosi fungoide follicolare (più comune nei pazienti anziani) [1].
I depositi di mucina nel contesto delle malattie multisistemiche
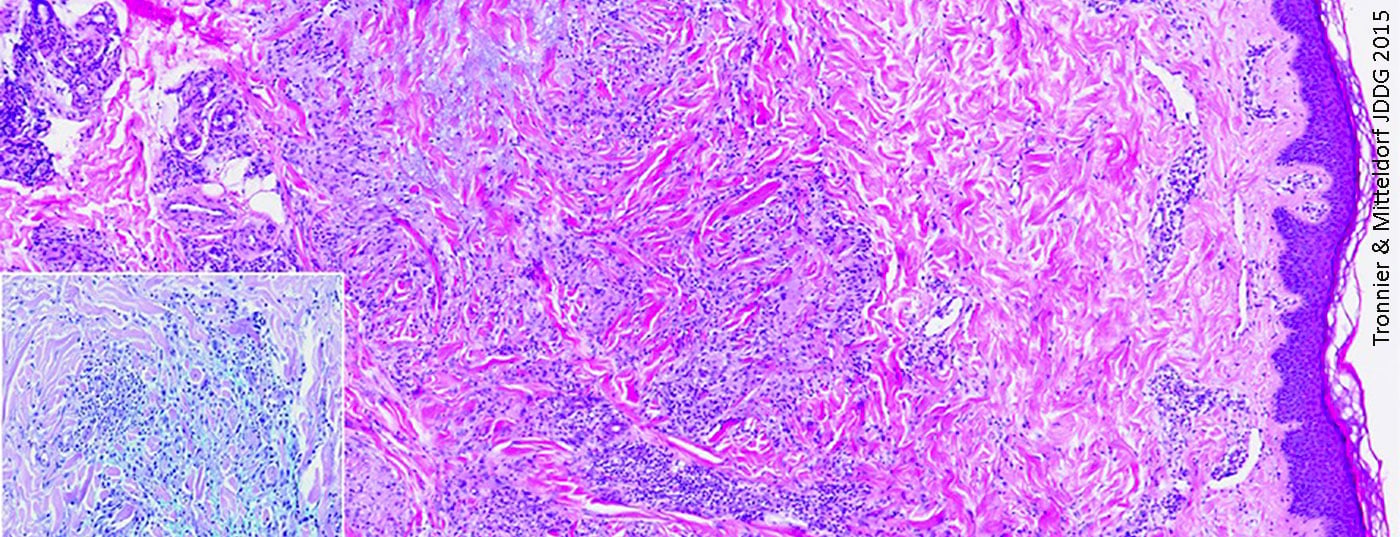
Il granuloma anulare (Fig. 1) è una malattia granulomatosa non infettiva di eziologia sconosciuta [6], che si verifica frequentemente nei pazienti più giovani [7]. I siti di predilezione per i sintomi, che di solito si manifestano sotto forma di placche anulari o arciformi, sono principalmente le estremità [7]. La deposizione di mucina nell’area di degenerazione del tessuto connettivo è una caratteristica istologica importante [8]. Questo è facilmente riconoscibile a livello istopatologico nelle macchie di blu di Alcian o di ferro colloidale [8]. Nella maggior parte dei casi, l’epidermide non è colpita; nel derma c’è tessuto connettivo degenerato, circondato da cellule epitelioidi disposte in modo leggermente allungato e simile a una palizzata [8]. Oltre alla forma solitaria, esiste anche una forma disseminata, che può verificarsi, ad esempio, nel contesto del diabete, che colpisce soprattutto i casi in età avanzata [7].

Il lupus eritematoso è una malattia infiammatoria autoimmune con manifestazioni cliniche e decorso eterogenei [9]. Il lupus eritematoso sistemico è una malattia multisistemica potenzialmente pericolosa per la vita, con coinvolgimento cutaneo. La comparsa di lesioni cutanee durante il decorso di questa malattia è comune e può verificarsi fino al 70-85% dei pazienti [9]. Nel lupus eritematoso cutaneo acuto, si distingue una forma localizzata (eritema a farfalla) da una variante generalizzata (esantema maculopapulare), che è il sottotipo più comune del lupus eritematoso sistemico (30-60%) [9]. Dal punto di vista istopatologico, una dermatite di interfaccia povera di cellule e un infiltrato linfocitario perivascolare piuttosto rado con depositi di mucina sono i risultati tipici del lupus eritematoso cutaneo acuto [9].
La dermatomiosite è un’altra malattia primaria con depositi di mucina cutanea come caratteristica istopatologica tipica [10]. Questi si verificano nel contesto di focolai infiammatori nell’area dello strato di transizione dermo-epidermico [10].
Fonte: ZDFT, Zurigo
Letteratura:
- Dummer R, Guillet C, Nordmann Th: Presentazione di diapositive: tema annuale – dermatosi deposizionali. Mucinosi. Prof. Reinhard Dummer & Team, 9° Giornate di formazione dermatologica 2019, Zurigo, 26 giugno 2019.
- Rongioletti F, Rebora A: Le mucinosi cutanee: criteri microscopici per la diagnosi. Am J Dermatopathol 2001; 23: 257-267.
- Kuhn A: Mucinosi. Dermatologia, Venereologia e Allergologia di Braun-Falco 2017; 1-9
https://link.springer.com/referenceworkentry/10.1007%2F978-3-662-49546-9_93-1) - Schallera J, Meigelb WN: Dermatosi da deposito, in: Istopatologia della pelle ed: Cerroni L et al, 2a edizione 2016, Springer: Berlin Heidelberg. DOI 10.1007/978-3-662-44367-5_24-1, https://link.springer.com/content/pdf/10.1007%2F978-3-662-44367-5_24-1.pdf
- Rongioletti F, et al: Mucinosi eritematosa reticolare: revisione delle caratteristiche dei pazienti, condizioni associate, terapia ed esito in 25 casi. Br J Dermatol 2013; 169: 1207-1211.
- Piette EW, Rosenbach M: Granuloma annulare. Patogenesi, associazioni e fattori scatenanti della malattia e opzioni terapeutiche. JAAD 2016; 75 (3): 467-479. www.jaad.org/article/S0190-9622(15)01500-5/abstract
- Houriet C, et al.: Hautmanifestationen bei inneren Erkrankungen – 1.Teil, Universitätsklinik für Dermatologie, Inselspital Bern. Schweiz Med Forum 2013;13(47): 949-953, https://boris.unibe.ch/45902/1/houriet_smf_1.pdf
- Tronnier M, Mitteldorf C: Caratteristiche istologiche delle malattie cutanee granulomatose: Parte 1: Malattie granulomatose non infettive. Minireview. JDDG 2015; 13 (3): 211-216, https://onlinelibrary.wiley.com/doi/full/10.1111/ddg.12610_suppl
- Sticherling M, Kuhn A: Lupus eritematoso. Dermatologia, Venereologia e Allergologia di Braun-Falco 2017; 1-18, https://link.springer.com/referenceworkentry/10.1007%2F978-3-662-49546-9_54-1
- Miller ML: Diagnosi e diagnosi differenziale della dermatomiosite e della polimiosite negli adulti. UpToDate, www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-dermatomyositis-and-polymyositis-in-adults
DERMATOLOGIE PRAXIS 2019; 29(5): 41-43 (pubblicato il 10.10.19, prima della stampa).









