Lo stato epilettico è definito come una crisi epilettica con una durata di >5 min o una serie di crisi tra le quali non viene recuperato lo stato neurologico originale. Una delle opzioni più importanti per il trattamento primario dell’epilessia di stato è la somministrazione rapida e adeguata di benzodiazepine, disponibili in varie forme.
Può sostenere il test ECM nella nostra piattaforma di apprendimento dopo aver esaminato i materiali consigliati. Clicchi sul seguente pulsante:
Lo stato epilettico è definito come una crisi epilettica della durata di >5 minuti o una serie di crisi tra le quali non viene recuperato lo stato neurologico originale. Con un’incidenza di 10-40 per 100.000 anni-persona e un tasso di mortalità compreso tra il 7 e il 33%, lo stato epilettico (SE) è una delle emergenze neurologiche più comuni, ma è anche una condizione acutamente pericolosa per la vita [1,2]. Con l’8,2%, la mortalità nei pazienti senza coscienza compromessa è significativamente più bassa rispetto ai pazienti con coscienza compromessa (33%) [2]. Le conseguenze a lungo termine possono includere disturbi neurologici, cognitivi e comportamentali e una riduzione significativa della qualità della vita. Inoltre, l’epilessia di stato può essere associata a un significativo deterioramento dell’esito clinico. Complicazioni come fratture, immobilità, polmonite da aspirazione acquisita come parte dello status, ma anche una limitazione delle abilità della vita quotidiana come risultato di un processo di recupero prolungato dopo la congestione o la perdita di funzionalità dovuta a una lunga permanenza nel reparto di terapia intensiva giocano un ruolo in questo caso. L’esito dopo l’epilessia di stato è determinato principalmente dall’eziologia dell’epilessia di stato, dal tipo o dallo stadio dell’epilessia di stato, dalla sua durata e anche dall’età del paziente [3]. I predittori per la SE ricorrente erano l’età <4 anni, il sesso femminile, la mancanza di responsività al farmaco alla prima dose e le eziologie sintomatiche e progressive.
Terapia conforme alle linee guida dell’epilessia di stato
Il successo del trattamento della SE è critico in termini di tempo e dipende dalla tempestività della terapia e della diagnosi neurologica. La fase preclinica è quindi di grande importanza per l’erogazione delle cure. Il trattamento della SE negli adulti è organizzato in uno schema a tappe nella linea guida attuale (Tabella 1) [4]. Dopo le misure generali, come assicurare i segni vitali (schema ABCDE), proteggere la testa dalle lesioni, somministrare O2 a una saturazione di O2 di <95% e una riduzione sintomatica della temperatura a >37,5°C, la terapia iniziale (fase 1) consiste nella somministrazione di benzodiazepine. Le dosi iniziali negli adulti >40 kg di peso corporeo (bw) sono: Lorazepam 0,1 mg/kg di peso corporeo (massimo 4 mg/bolus, ripetere una volta se necessario) o clonazepam 0,015 mg/kg di peso corporeo (massimo 1 mg/bolus, ripetere una volta se necessario) o midazolam 0,2 mg/kg di peso corporeo (massimo 1 mg/bolus, ripetere una volta se necessario). 10 mg/bolus per via intramuscolare (i.m.), endovenosa (i.v.) o intranasale (i.n.), ripetere 1× se necessario) o diazepam 0,15-0,2 mg/kg di peso corporeo (massimo 10 mg/bolus per somministrazione, ripetere 1× se necessario). Nei pazienti senza accesso endovenoso, il midazolam deve essere somministrato per via intramuscolare tramite applicatore o per via intranasale (10 mg per >40 kg, 5 mg per <40-13 kg di peso corporeo) come dose singola). Se la SE persiste dopo la somministrazione iniziale di una benzodiazepina, si deve verificare se la dose era adeguata, poiché il sottodosaggio della terapia iniziale è comune a causa della paura degli effetti collaterali inerenti alle benzodiazepine e può comportare un ridotto controllo delle crisi. Se necessario, la benzodiazepina deve essere somministrata nuovamente in una dose sufficientemente elevata come parte della terapia iniziale.
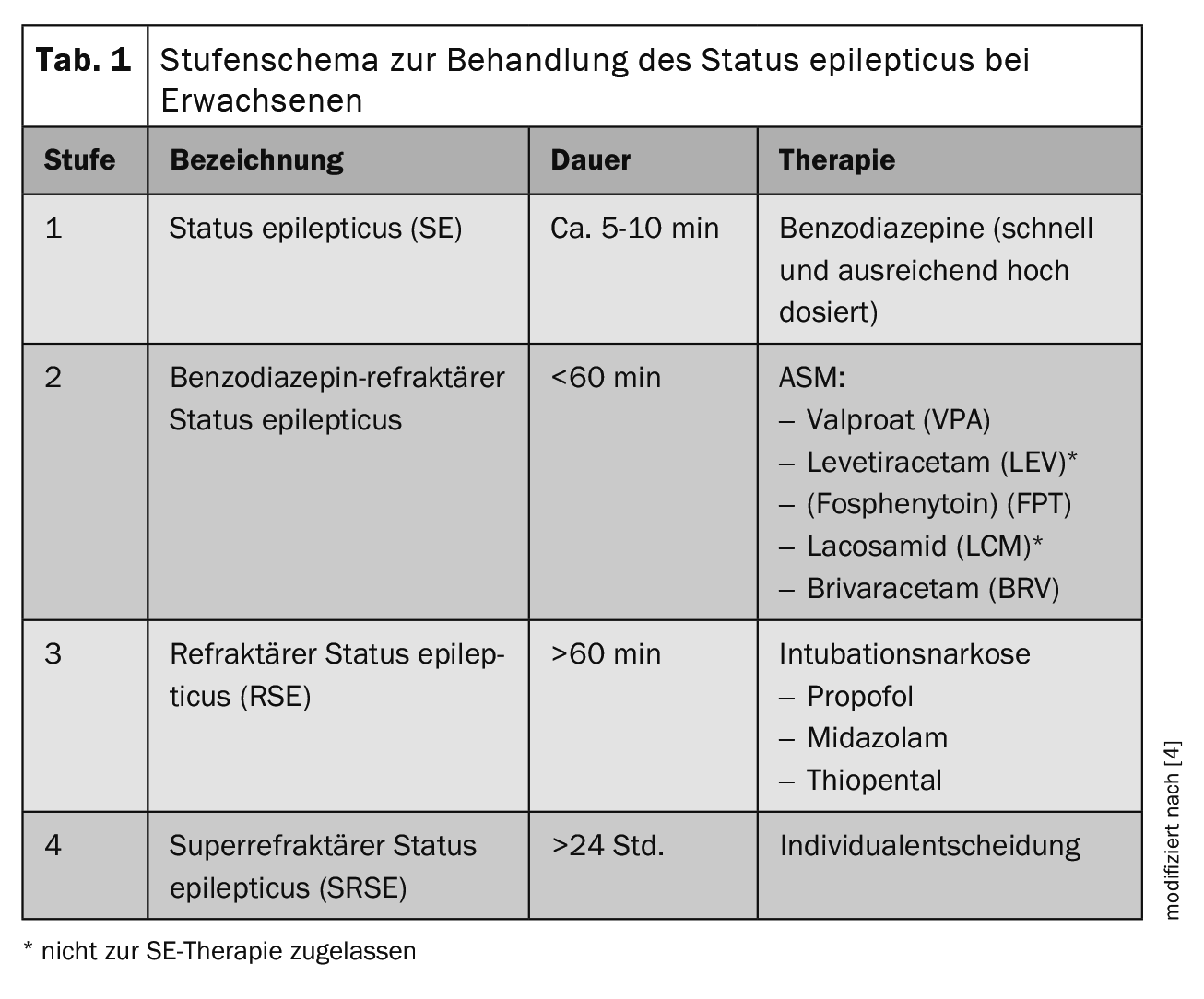
Se la dose iniziale di benzodiazepina era sufficientemente alta, il paziente dovrebbe essere in grado di assumerla entro 30 minuti. nella Il secondo livello di terapia è costituito dai soppressori di convulsioni disponibili per via endovenosa (ASM). Come medicinali della Levetiracetam (LEV, 60 mg/kg di peso corporeo, massimo 4500 mg per >10 min i.v.) (non approvato per la terapia della SE), valproato (VPA, 40 mg/kg di peso corporeo, massimo 3000 mg per >10 min i.v.) o fosfenitoina (FPHT, 20 mg/kg di peso corporeo, massimo 1500 mg per >10 min i.v.) devono essere somministrati come prima scelta [4]. Sebbene la fosfenitoina sia autorizzata in Germania e in Austria, non è commercializzata in questi Paesi e non è autorizzata in Svizzera, quindi questo non è rilevante nell’attuazione pratica della terapia nei Paesi di lingua tedesca. Un’altra possibile alternativa è la somministrazione di lacosamide in una dose di 5 mg/kg i.v., che può essere somministrata in 15 minuti [5]. Tuttavia, va notato che una controindicazione nel caso di un blocco AV 2. o Il 3° grado esiste. Anche in questo caso, non esiste un’autorizzazione per la terapia SE. Le prime serie di casi e le relazioni sui casi descrivono il successo dell’uso di brivaracetam somministrato per via endovenosa nella SE refrattaria [6].
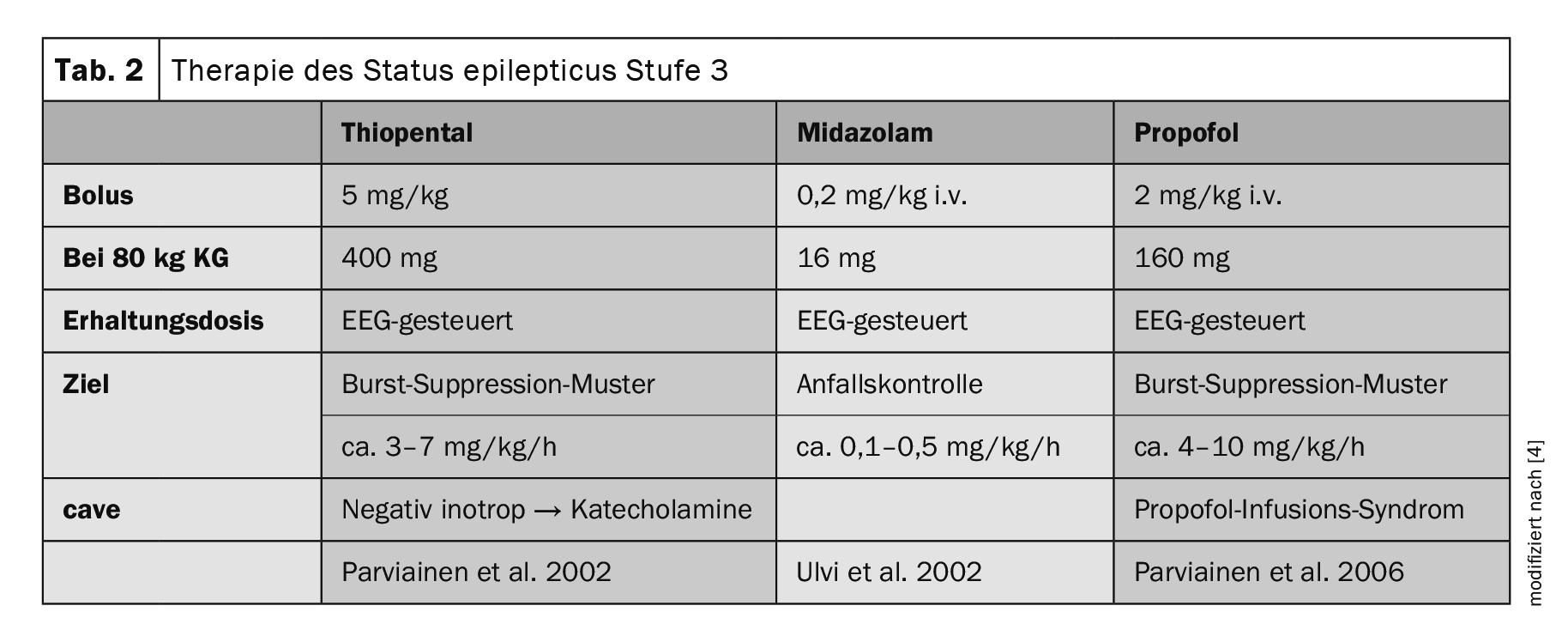
Dopo il trattamento di fase 2, l’anestesia per intubazione con tiopentale, midazolam o propofol viene eseguita se l’ASM endovenosa fallisce (Tabella 2) [5]. Se anche questo livello di terapia dovesse fallire, la linea guida attuale suggerisce ulteriori opzioni di trattamento, anche se queste si basano in gran parte su rapporti di casi individuali. A questo punto, la SE viene definita super refrattaria (SRSE). Oltre alla somministrazione di barbiturici, ketamina, antagonisti NMDA, anestetici per inalazione come l’isoflurano o il desflurano, si può prendere in considerazione la somministrazione enterale di altri ASM “classici”, disponibili solo per via orale, oppure prove di terapia individuale con lidocaina, dieta chetogenica e chirurgia dell’epilessia.
Problemi attuali nel trattamento dell’epilessia di stato
Il trattamento rapido e a dosi sufficientemente elevate della SE è di grande importanza prognostica. La terapia iniziale, in particolare, spesso si discosta dalle raccomandazioni della linea guida. Guterman et al. 2021 sono stati in grado di dimostrare che il trattamento della SE preclinica raramente è conforme alle linee guida specificate. Dei 9176 ricoveri pre-ospedalieri per stato epilettico in 743 strutture, 7665 pazienti (83,6%) sono stati trattati con midazolam, 1264 (13,8%) con lorazepam e 245 (2,7%) con diazepam. Ci sono stati 357 (3,9%; 95% CI: 3,5%-4,3%) casi in cui il trattamento iniziale era in linea con le raccomandazioni degli esperti in termini di dose e tipo raccomandato. La maggior parte dei pazienti ha quindi ricevuto dosi inferiori di benzodiazepine rispetto a quelle raccomandate [7].
Nello studio SENSE, uno studio di coorte trinazionale, Kellinghaus et al. hanno riportato che nel 15% di tutti i casi non è stata somministrata alcuna benzodiazepina nella prima fase del trattamento. I dati hanno dimostrato che l’uso conforme alla linea guida delle benzodiazepine era associato al successo del trattamento e a un numero significativamente più elevato di SE da sfondamento [8].
In sintesi, tutti gli studi hanno dimostrato che la somministrazione preospedaliera di benzodiazepine abbrevia il tempo di controllo delle crisi e riduce la durata del ricovero in ospedale nei pazienti con SE.
Per ottimizzare la terapia iniziale, la somministrazione semplificata delle benzodiazepine attraverso l’approvazione di iniettori e applicazioni nasali/buccali rappresenta una semplificazione della somministrazione iniziale da parte dei soccorritori e degli assistenti domiciliari, in modo che la terapia iniziale possa essere ulteriormente ottimizzata in futuro [9].
Gawedzki et al. sono riusciti a dimostrare nel 2022, in uno studio osservazionale retrospettivo e monocentrico nel reparto di emergenza, che la presenza di un farmacista accompagnatore nell’équipe dello status epilettico riduceva il tempo mediano di somministrazione del farmaco. 1. e Il secondo ASM è stato ridotto in modo significativo. Inoltre, il gruppo di pazienti con la presenza di un farmacista ha ricevuto una dose mediana più alta di lorazepam equivalente (2,5 mg [IQR 2–4] vs. 2 mg [IQR 2–2]; p=0,04) e ha avuto più probabilità di ricevere una dose iniziale sufficientemente alta di almeno 4 mg di lorazepam equivalente (38% vs. 0%; p=0,11). Tuttavia, non ci sono state differenze nella durata della degenza ospedaliera o nella mortalità a 30 giorni [10]. Da ciò si è concluso che la presenza di un farmacista o di un osservatore della terapia aumenta la consapevolezza della terapia conforme alle linee guida.
Un altro fattore clinicamente rilevante è il riconoscimento tempestivo dell’epilessia di stato non convulsa (NCSE), in quanto il ritardo nella diagnosi e nel trattamento della SE comporta un aumento della mortalità. La SE non convulsiva è una delle emergenze neurologiche più frequentemente trascurate, anche perché spesso è associata a gravi patologie interne che rendono difficile la diagnosi [11]. Lo studio di incidenza di Leitinger et al. ha mostrato che la SE non convulsa è associata a un alto tasso di mortalità (CFR 27,65%). Con un’incidenza di 12,1/100.000, è un’emergenza comune in epilettologia [2].
La NCSE/NCS (crisi non convulsiva) è stata rilevata nel 21% dei 170 soggetti ricoverati in un’unità di terapia intensiva. Le crisi cliniche hanno preceduto la diagnosi EEG di NCSE/NCS solo nel 25% dei casi. I principali fattori di rischio per la NCSE/NCS sono stati precedenti malattie del SNC, ad esempio tumori del SNC, epilessia nota, meningite/encefalite o evidenza di encefalomalacia alla risonanza magnetica [12,13].
Se lo stato epilettico persiste anche dopo 1 ora o 24 ore di terapia, si parla di SE refrattaria o sovra-refrattaria. La SE refrattaria e sovra-refrattaria ha un esito significativamente peggiore rispetto allo status epilettico non complicato. Strzelczyk et al. ha analizzato retrospettivamente l’esito e la durata del ricovero dei pazienti con epilessia di stato refrattaria e super-refrattaria. A tale scopo, è stato utilizzato il database “Gesundheitsforen Leipzig”, che conteneva diagnosi, costi e dati demografici di pazienti con SE, ricoverati e ambulatoriali. La percentuale maggiore di pazienti con SE non refrattaria è stata dimessa a casa (78,1%), mentre questo valeva per il 70,1% dei pazienti con RSE e solo per il 31,7% dei pazienti con SRSE. Più di un terzo dei pazienti con un SRSE (39,9%) è morto rispetto al 15% dei pazienti RSE e al 9,6% dei pazienti nRSE [14].
Nuovi approcci terapeutici per la terapia della SE sovrarefrattaria
Attualmente si stanno discutendo approcci terapeutici nuovi e già noti come possibili opzioni di trattamento per l’epilessia di stato refrattaria al trattamento. Prima di tutto, è necessario menzionare ulteriori tentativi di terapia farmacologica:
In uno studio di coorte bicentrico svizzero, sono stati inclusi 205 pazienti, il 27% dei quali è stato sottoposto ad anestesia dopo il fallimento del farmaco di prima linea. I risultati hanno mostrato che l’anestesia come trattamento di seconda linea era associata a una durata mediana della SE più breve (0,5 contro 12,5 giorni, p<0,001), a un tempo più breve in terapia intensiva (2 contro 5,5 giorni, p<0,001) e a una durata ridotta della degenza ospedaliera (8 contro 17 giorni, p<0,001) con tassi uguali di complicazioni rispetto all’anestesia come trattamento di terza linea. [15].
Recentemente, è stato discusso l’uso del fenobarbital per il trattamento della SRSE. Il fenobarbital è uno dei più vecchi ASM, essendo in uso clinico dal 1912. Ci sono vari resoconti di un forte effetto soppressivo delle crisi epilettiche, con poca sedazione. Allo stesso tempo, durante il trattamento con fenobarbital sono stati segnalati possibili effetti collaterali come ipotensione, aritmie, aumento del tasso di infezioni e ipopnea. Il fenobarbital determina un aumento dell’inibizione GABAergica e una riduzione dell’eccitazione glutammatergica, oltre all’inibizione dei recettori AMPA. In singoli casi, il fenobarbital sembra essere un’opzione terapeutica per il trattamento dell’RSE che non dovrebbe essere dimenticata. Come per altri ASM, sarebbe auspicabile e necessario un RCT per valutare l’importanza nel trattamento della SE [16].
Le opzioni terapeutiche non farmacologiche discusse nella linea guida attuale includono le seguenti: Stimolazione transcranica a corrente diretta (tDCS), stimolatore del nervo vago (VNS), dieta chetogenica e raffreddamento focale:
La TDCS è una tecnica di neuromodulazione non invasiva che applica una debole stimolazione di corrente diretta attraverso il cuoio capelluto per indurre effetti polarizzati lineari e non lineari. In particolare, la stimolazione catodica induce un’iperpolarizzazione nelle cellule neuronali e provoca effetti acuti e a lungo termine potenzialmente rilevanti nella fisiopatologia della SRSE. Ng et al. ha testato la fattibilità dell’uso della stimolazione transcranica a corrente diretta ad alta definizione (hd-tDCS) nel trattamento dell’RSE. In 10 pazienti RSE, non si sono verificati eventi avversi in 32 sessioni di hd-tDCS. La TDCS potrebbe essere potenzialmente associata a una riduzione acuta dell’input presinaptico eccitatorio o alla depressione della forza sinaptica mediata dai recettori N-metil-D-aspartato (NMDA), che possono produrre effetti di lunga durata, tra cui l’azione transmembrana, la migrazione delle proteine e/o gli effetti antinfiammatori [17].
Il trattamento della SE sopra-refrattaria con la dieta chetogenica (KD) è un approccio molto promettente. In uno studio di coorte retrospettivo non randomizzato, Koh et al. 140 pazienti RSE. Questo include 32 pazienti che sono stati trattati con una KD. Di questi, la SE è stata interrotta in 28 casi (81%). L’uso del KD ha influenzato la riduzione della scala di classificazione modificata (mRS) alla dimissione, nei pazienti più anziani, con punteggi di gravità delle crisi più elevati, sotto terapia di anestesia continua per via endovenosa (CIVAD) e nei pazienti con SRSE. Inoltre, l’età e i punteggi di gravità delle crisi, ma non il CIVAD o l’SRSE, erano associati a un cambiamento mediato dalla KD nel punteggio mRS a 3 mesi. Sulla base di questi dati, gli autori discutono di un possibile effetto neuroprotettivo della KD nei pazienti SRSE [18].
Come metodo altamente sperimentale, Niesvizky-Kogan et al. ha recentemente introdotto il principio del raffreddamento focale come opzione terapeutica per le epilessie refrattarie e la SE. Durante il raffreddamento, il rilascio di neurotrasmettitori dalla presinapsi è ridotto e le spine dendritiche nella postsinapsi vengono perse. Inoltre, devono essere influenzate le proprietà elettriche, gli acidi nucleici, i neurotrasmettitori e la funzione dei canali della membrana cellulare. Questo è analogo al raffreddamento globale discusso nel contesto della neuroprotezione nell’arresto post-cardiovascolare e nella lesione ischemica neonatale, ma è discusso come un metodo più sicuro [19].
L’epilessia di stato refrattaria di nuova insorgenza (NORSE) rappresenta una sfida per la terapia. La maggior parte dei pazienti con epilessia di stato refrattaria di nuova insorgenza sviluppa un SRSE con un decorso clinicamente sfavorevole e un tasso di mortalità del 12-27% [3]. Come Sculier et al. come descritto in una revisione, la terapia è spesso difficile, con il 75% dei pazienti NORSE che richiedono una terapia anestetica. Gli autori sono riusciti a dimostrare che l’encefalite autoimmune è la causa di una buona metà dei casi, per cui si raccomanda una terapia immunosoppressiva precoce, come il prednisolone o, se necessario, le immunoglobuline per via endovenosa e la separazione del plasma o, come terapia di seconda linea, i farmaci immunomodulatori come il rituximab [3]. I pazienti con NORSE devono anche ricevere inizialmente una terapia conforme alle linee guida per la SE. Se nel corso della malattia si dovesse sviluppare un decorso refrattario della SE appena insorta, Sculier et al. immunoterapia entro le prime 72 ore.
Un altro approccio terapeutico interessante è la modulazione dei fattori che influenzano lo stato epilettico, come il metabolismo del glucosio e i livelli di piridossal fosfato. In uno studio di coorte retrospettivo monocentrico di Müller et al. Lo studio ha analizzato se le complicazioni del trattamento endovenoso con valproato, che viene utilizzato per trattare la SE, sono diverse nei pazienti con o senza diabete. Durante il periodo di studio, 408 pazienti e 482 episodi di SE sono stati trattati per via endovenosa con VPA. I confronti tra i gruppi non hanno mostrato differenze significative nei tassi di interruzione del trattamento. Sono state riscontrate differenze nel tasso di trombocitopenia (p=0,015), che si è verificata più frequentemente nei pazienti con diabete. Sono stati identificati un totale di 36 episodi ipoglicemici, due dei quali si sono verificati spontaneamente sotto VPA. Gli autori sono giunti alla conclusione che il diabete, in quanto comorbilità rilevante, comporta un rischio potenzialmente maggiore di esito negativo dopo la SE [20].
Uno studio di coorte retrospettivo di Rubinos et al. con un totale di 293 pazienti, ha studiato la relazione tra i livelli di piridossal fosfato (PLP) e la SE accertata (eSE). Il livello mediano di PLP del gruppo eSE (12 nmol/l) era inferiore a quello del gruppo ICU-noSE (22 nmol/l, p=0,003), del gruppo fuori dall’ICU (16 nmol/l, p=0,05) e del gruppo ambulatoriale (36 nmol/l, p <0,001). I pazienti con eSE avevano quindi una prevalenza significativamente più alta di livelli marginali e ridotti di PLP (90 e 80%, rispettivamente) rispetto agli altri pazienti in terapia intensiva e non in terapia intensiva (ICU-noSE: 70, 50%; non-ICU: 63, 54%; pazienti ambulatoriali: 38, 21%) [21]. Tuttavia, mancano ancora studi terapeutici sulla somministrazione di PLP.
Conclusione
Lo stato epilettico è una delle emergenze più comuni in neurologia. La progressione dell’epilessia di stato refrattaria e super-refrattaria, in particolare, continua a rappresentare una sfida per la pratica clinica. Una dose sufficientemente alta di terapia iniziale, somministrata il prima possibile dopo la diagnosi, può ridurre il tasso di corsi refrattari. Lo stato epilettico, soprattutto se la coscienza non viene mantenuta, è una condizione potenzialmente fatale che inizialmente richiede un trattamento medico e un monitoraggio intensivo. La terapia conforme alle linee guida deve essere fornita fin dall’inizio. Si dovrebbero prendere in considerazione nuovi approcci terapeutici per il trattamento dell’RSE, come la dieta chetogenica o la tDCS, che hanno mostrato risultati iniziali promettenti. Sono necessari ulteriori studi per valutare l’efficacia al di fuori dei singoli casi.
Messaggi da portare a casa
- Lo stato epilettico è definito come una crisi epilettica della durata di >5 minuti o una serie di crisi tra le quali non viene recuperato lo stato neurologico originale.
- Una delle opzioni più importanti per il trattamento primario dell’epilessia di stato è la somministrazione rapida e adeguata di benzodiazepine, che sono disponibili in varie forme (endovenosa, intramuscolare, intranasale, buccale/sublinguale, rettale).
- L’epilessia di stato deve essere trattata in un’unità di terapia intensiva.
Letteratura:
- Knake S, Rosenow F, Vescovi M, et al: Gruppo di Studio sullo Stato Epilettico dell’Assia (SESGH). Incidenza dell’epilessia di stato negli adulti in Germania: uno studio prospettico basato sulla popolazione. Epilepsia. 2001 Jun; 42(6): 714-718. doi: 10.1046/j.1528-1157.2001.01101.x. PMID: 11422324.
- Leitinger M; Trinka E; Giovannini G, et al: Epidemiologia dell’epilessia di stato negli adulti: uno studio basato sulla popolazione su incidenza, cause ed esiti (2019). In: Epilepsia 60(1), 53-62. doi: 10.1111/epi.14607.
- Sculier C, Gaínza-Lein M, Sánchez Fernández I, Loddenkemper T: Esiti a lungo termine dell’epilessia di stato: una valutazione critica (2018). In: Epilepsia 59 Suppl 2, 155-169. doi 10.1111/epi.14515.
- Rosenow F, Weber J, et al: Epilessia di stato negli adulti. Linea guida S2k. (2020): A cura della Società Tedesca di Neurologia (DGN). Disponibile online su www.dgn.org/leitlinien, ultimo aggiornamento il 30.06.2020, ultima revisione il 27.10.2023.
- Misra Usha K, Dubey D, Kalita J: Uno studio randomizzato controllato di lacosamide rispetto al valproato di sodio nello status epilettico (2017). In: Epilepsia. doi: 10.1111/epi.13706.
- Strzelczyk A, Steinig I, Willems LM, et al: Trattamento dell’epilessia di stato refrattaria e super-refrattaria con brivaracetam: uno studio di coorte di due ospedali universitari tedeschi. (2017b). In: Epilessia e comportamento: E&B 70 (Pt A), 177-181. doi: 10.1016/j.yebeh.2017.03.028.
- Guterman EL, Burke JF, Sporer KA: Trattamento preospedaliero dello stato epilettico negli Stati Uniti (2021). In: JAMA 326 (19), 1970-1971. doi: 10.1001/jama.2021.15964.
- Kellinghaus C, Rossetti AO, Trinka E, et al: Registro SENSE per lo status epilettico. Epilepsia. 2018 Oct; 59 Suppl 2: 150-154.
doi: 10.1111/epi.14495. Epub 2018 Aug 29. PMID: 30159884 - Halliday AJ, Santamaria J, D’Souza WJ: Le benzodiazepine pre-ospedaliere associate a un miglioramento degli esiti nell’epilettico di stato extraospedaliero: uno studio di coorte retrospettivo di 10 anni (2021). In: Epilepsy research 179, 106846. doi: 10.1016/j.eplepsyres.2021.106846.
- Gawedzki P, Celmins L, Fischer D: Coinvolgimento del farmacista nella terapia antiepilettica per lo status epilettico nel dipartimento di emergenza (2022). In: The American journal of emergency medicine 59, 129-132. doi: 10.1016/j.ajem.2022.07.002.
- Drislane FW: Presentazione, valutazione e trattamento dell’epilessia di stato non convulsiva (2000). In: Epilessia e comportamento: E&B 1 (5), 301-314. doi: 10.1006/ebeh.2000.0100.
- Laccheo I, Sonmezturk H, Bhatt AB, et al: Stato epilettico non convulsivo e crisi non convulsive in pazienti neurologici in terapia intensiva (2015). In: Neurocritical care 22 (2), 202-211. doi: 10.1007/s12028-014-0070-0.
- Spindler M, Jacks LM, Chen X, et al: Spettro dell’epilessia di stato non convulsa nei pazienti con cancro (2013). In: Journal of clinical neurophysiology: official publication of the American Electroencephalographic Society 30 (4), 339-343. doi: 10.1097/WNP.0b013e31829ddcdb.
- Strzelczyk A, Ansorge S, Hapfelmeier J, et al: Costi, durata della degenza e mortalità dell’epiletto di stato super-refrattario: uno studio basato sulla popolazione della Germania (2017a). In: Epilepsia 58 (9), 1533-1541. DOI: 10.1111/epi.13837.
- Sutter R, Jünger AL, Baumann SM, et al: Bilanciare i rischi e i benefici degli anestetici nello status epilettico (2023). In: Epilessia & comportamento: E&B 138, 109027. doi: 10.1016/j.yebeh.2022.109027.
- Trinka E: Il fenobarbital nello stato epilettico – Riscoperta di un farmaco efficace. Epilepsy Behav 2023 Apr; 141: 109104. doi: 10.1016/j.yebeh.2023.109104.
- Ng MC, El-Alawi H, Toutant D, et al: A Pilot Study of High-Definition Transcranial Direct Current Stimulation in Refractory Status Epilepticus: The SURESTEP Trial (2023). In: Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics 20(1), 181-194. doi: 10.1007/s13311-022-01317-5.
- Koh S, Kim T-J, Shin H-B, et al.: Expanding Indications for a Ketogenic Diet as an Adjuvant Therapy in Adult Refractory Status Epilepticus: an Exploratory Study Using Moderation Analysis (2022). In: Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics 19 (5), 1526–1534. doi: 10.1007/s13311-022-01282-z.
- Niesvizky-Kogan I, Bass M, Goldenholz SR, Goldenholz M: Raffreddamento focale per l’epilessia resistente ai farmaci: una revisione (2022). In: JAMA neurology 79 (9), 937-944. doi: 10.1001/jamaneurol.2022.1936.
- Müller A, Hofen-Hohloch Jv, Awissus C, et al: Il diabete mellito influenza il profilo di sicurezza dell’acido valproico per il trattamento dell’epilessia di stato? Uno studio retrospettivo di coorte (2022). In: Ricerca e pratica neurologica 4(1), 52. doi: 10.1186/s42466-022-00212-w.
- Rubinos C, Bruzzone MJ, Blodgett C, et al: Associazione dei livelli sierici di piridossal fosfato con lo stato epilettico accertato (2023). In: Neurocritical care 38(1), 41-51. DOI: 10.1007/s12028-022-01579-z.
HAUSARZT PRAXIS 2024; 19(9): 12–16