La sarcoidosi può manifestarsi a qualsiasi età e colpisce tipicamente i polmoni, ma possono essere coinvolti anche altri organi. La diagnosi consiste in diverse fasi; non esistono esami del sangue specifici che confermino o escludano la sarcoidosi. Istologicamente, la sarcoidosi è caratterizzata da granulomi cellulari epitelioidi, non necrotizzanti. Una volta identificata la sarcoidosi polmonare, è importante cercare un coinvolgimento extratoracico rilevante; le manifestazioni degli organi cardiaci, in particolare, possono essere pericolose per la vita.
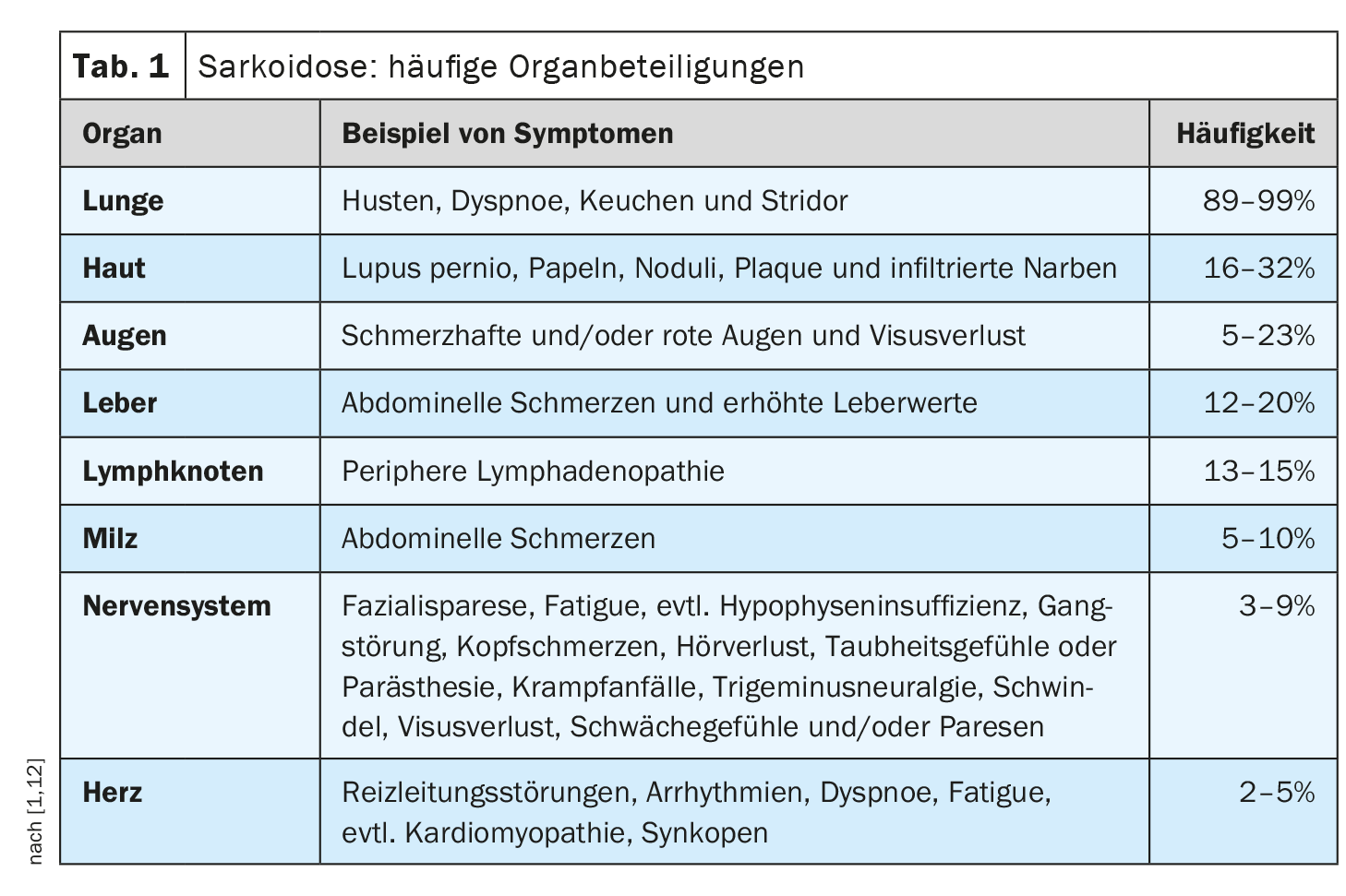
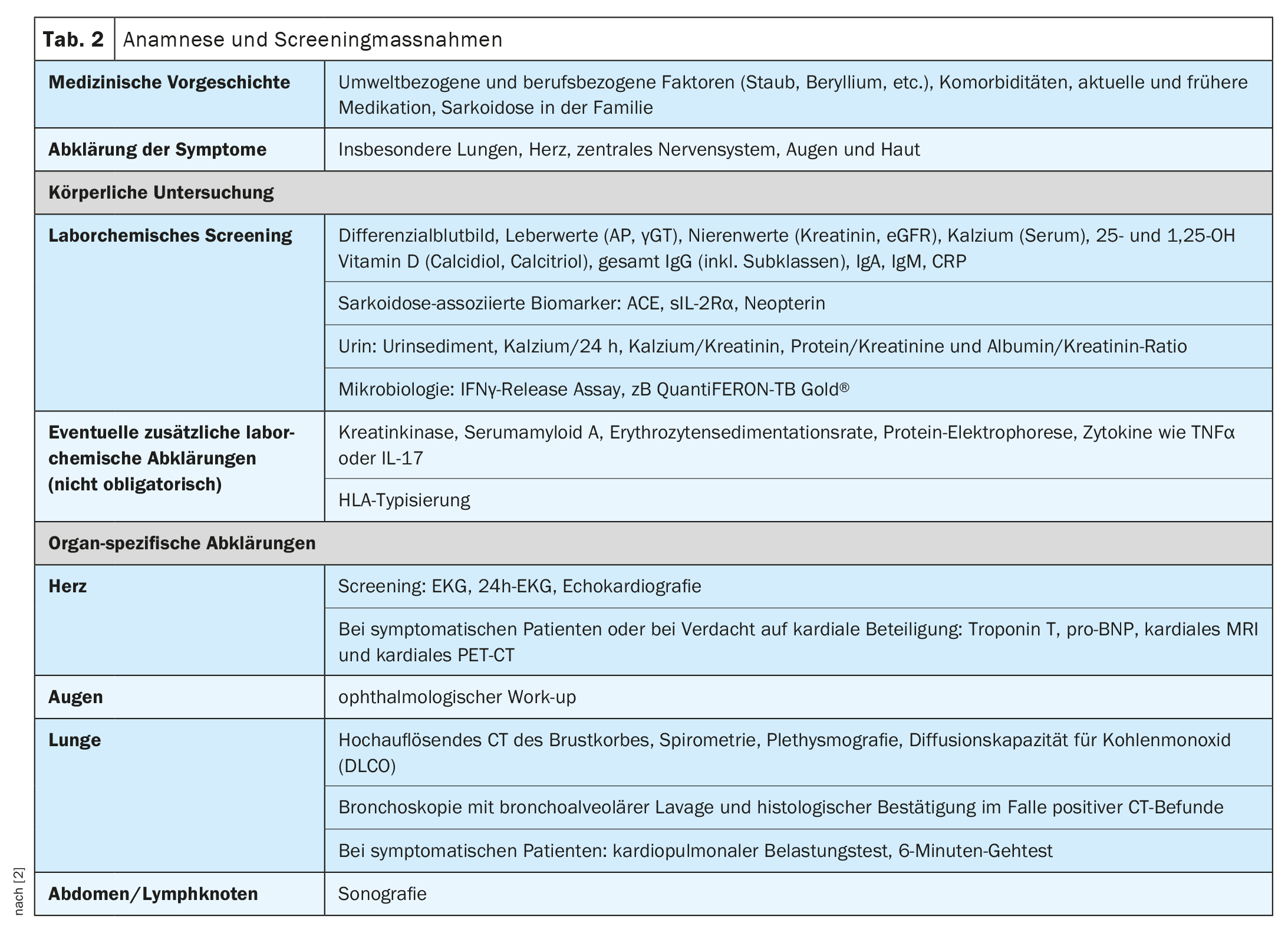
Il decorso clinico e la prognosi della sarcoidosi sono molto eterogenei e dipendono dal rispettivo coinvolgimento degli organi, ha spiegato il Prof. Dr med. Jörg D. Seebach, medico chef del Service d’immunologie et allergologie, Hôpitaux universitaires de Genève [1]. I polmoni, i linfonodi, la pelle e gli occhi sono i più comunemente colpiti, mentre le manifestazioni cardiache, renali e neurologiche sono meno comuni, ma sono associate a una maggiore morbilità [2]. La Tabella 1 offre una panoramica delle manifestazioni d’organo più comuni. [1,3]La diagnosi di sarcoidosi si basa principalmente sulle caratteristiche cliniche e radiologiche, sull’identificazione di granulomi non necrotizzanti in uno o più campioni di tessuto e sull’esclusione di altre cause di malattia granulomatosa. A volte la sarcoidosi è autolimitante – circa la metà dei pazienti con sarcoidosi sperimenta una remissione spontanea entro due anni e molti altri entro cinque anni dalla comparsa dei sintomi [4]. Tuttavia, esistono anche forme gravi di progressione. Le cause più comuni di morte nei pazienti con sarcoidosi sono l’insufficienza respiratoria nella fibrosi polmonare avanzata e il grave coinvolgimento cardiaco o neurologico [3]. La Tabella 2 riassume le raccomandazioni sull’anamnesi e le misure di screening e la Tabella 3 i chiarimenti in caso di sospetto coinvolgimento degli organi.
Le procedure broncoscopiche sono minimamente invasive.
Se si sospetta la sarcoidosi sulla base dei sintomi clinici e dei risultati radiografici, il relatore raccomanda di ordinare una biopsia [1]. In linea di principio, i campioni istologici devono essere prelevati dal sito che offre l’esposizione meno invasiva e le migliori possibilità diagnostiche. Poiché nella maggior parte dei casi sono interessati i polmoni, la broncoscopia è una procedura sicura e minimamente invasiva [2]. Esistono diverse procedure diagnostiche broncoscopiche, tra cui la biopsia endobronchiale (mucosa), la biopsia polmonare transbronchiale (TBLB) o l’agoaspirato transbronchiale (TBNA) dei linfonodi ilari/mediastinici e il lavaggio broncoalveolare (BAL). Sono possibili anche campioni biologici extrapolmonari, ad esempio dalla pelle, dalle ghiandole parotidi o lacrimali, dai linfonodi palpabili o dalle lesioni congiuntivali, ma meno specifici. In ogni caso, l’istologia deve essere accompagnata da manifestazioni cliniche e radiologiche compatibili e dall’esclusione di altre malattie. Se c’è una conferma istologica in siti extrapolmonari e un coinvolgimento polmonare concomitante con una sospetta infezione, come una malattia polmonare cavitaria, può essere necessaria una broncoscopia per escludere cause infettive come micobatteri e funghi [2].
La manifestazione polmonare è l’interessamento d’organo più comune.
Il coinvolgimento dei polmoni e/o dei linfonodi mediastinici/ilari è l’interessamento d’organo più comune, che si verifica in circa l’80-90% dei pazienti con sarcoidosi. La manifestazione polmonare è associata a un coinvolgimento parenchimale e a granulomi perivascolari. I sintomi più comuni sono una sensazione di pressione nel petto, tosse secca e dispnea. Nel decorso successivo, può svilupparsi una fibrosi con insufficienza respiratoria [5]. Poiché la sarcoidosi polmonare può presentarsi con modelli ostruttivi, restrittivi, misti o normali, i risultati dei test di funzionalità polmonare sono molto aspecifici, ma importanti per valutare la gravità, l’indicazione al trattamento e la risposta al trattamento. La malattia polmonare interstiziale (ILD) è un reperto tipico degli stadi 2, 3 e 4, che va dalle manifestazioni subcliniche alla fibrosi polmonare in fase terminale (stadio 4). Quest’ultima è un danno irreversibile agli organi, mentre l’ILD da lieve a moderata legata alla sarcoidosi è un sintomo trattabile.

La manifestazione cardiaca è meno frequente, ma prognosticamente sfavorevole.
Mentre circa il 90% di tutti i pazienti affetti da sarcoidosi presenta un coinvolgimento polmonare, la comparsa di sarcoidosi cardiaca è piuttosto rara, con una prevalenza del 2-7% [6]. Tuttavia, nei pazienti con sarcoidosi extracardiaca confermata, si raccomanda di ricercare il coinvolgimento cardiaco mediante un ECG [7]. Questo perché la sarcoidosi cardiaca è una manifestazione potenzialmente pericolosa per la vita [2]. Le moderne tecniche di imaging possono essere utili per la diagnosi precoce. Questi includono una risonanza magnetica cardiaca con tecnica di potenziamento del late-gadolinium (LGE) e FDG-PET [8]. Il coinvolgimento cardiaco può manifestarsi con aritmie ventricolari, blocco cardiaco di alto grado o insufficienza cardiaca cronica dovuta all’infiltrazione granulomatosa del miocardio e/o, nelle fasi più avanzate della malattia, alla fibrosi [2]. I possibili sintomi includono dolore al petto, palpitazioni, vertigini e sincope. La morte cardiaca improvvisa si verifica fino al 25%. [2,9]In sintesi, la diagnosi e il trattamento precoci del coinvolgimento cardiaco sono essenziali. [13]Le manifestazioni tipiche della sarcoidosi cardiaca sono disturbi della conduzione, aritmie ventricolari e insufficienza cardiaca. In circa un quarto dei casi, la sarcoidosi cardiaca si manifesta in modo isolato e senza coinvolgimento polmonare, secondo il Prof. Seebach [1]. Questo è spesso associato a una prognosi peggiore rispetto alla sarcoidosi sistemica con coinvolgimento cardiaco. Importanti diagnosi differenziali sono: miocardite linfocitaria, alcune cardiomiopatie genetiche, aumento fisiologico della captazione di FDG nell’insufficienza cardiaca [1].
Diagnosi differenziale: cosa bisogna considerare?
“La diagnosi differenziale è molto importante”, ha sottolineato il relatore [1]. I risultati istopatologici sono di grande importanza in questo contesto. La sarcoidosi è caratterizzata da granulomi compatti, non necrotizzanti, con una distribuzione perilinfatica lungo i fasci broncovascolari, parasettale e pleurica [2]. Nelle biopsie polmonari aperte, la sarcoidosi è spesso associata a vasculite granulomatosa senza distruzione delle pareti dei vasi. Nel corso della progressione della malattia, domina la fibrosi ialinizzata con resti di granulomi. La berilliosi cronica è una diagnosi differenziale essenziale alla sarcoidosi, soprattutto nei pazienti con esposizione ai metalli. Si deve considerare anche la malattia granulomatosa indotta da Infliximab. Queste entità possono essere ben differenziate istologicamente dalla sarcoidosi. Altre diagnosi differenziali includono tumori maligni (linfomi, carcinomi), collagenosi vascolari (lupus eritematoso sistemico, sindrome di Sjögren, cirrosi biliare primaria, artrite granulomatosa familiare), infezioni (HIV, [2,10,11] tubercolosi), vasculiti (granulomatosi con poliangioite, arterite di Takayasu, arterite a cellule giganti), polmonite da ipersensibilità, pneumoconiosi, malattia legata alle IgG4 e varie malattie da immunodeficienza). La morfologia e il modello di distribuzione dei granulomi correlati differiscono dalla sarcoidosi.
Le malattie infettive come la tubercolosi, il myobacterium avium, l’istoplasmosi, la coccidiomicosi e la malattia di Whipple) mostrano una distribuzione peribronchiale o casuale di granulomi e sono spesso associate a necrosi. L’uso di colorazioni speciali (Ziehl-Neelsen, auramina e argento) o di test PCR per il Mycobacterium tuberculosis complex e i miobatteri atipici può essere informativo per il rilevamento dei patogeni. A differenza della sarcoidosi, la polmonite da ipersensibilità è caratterizzata da accumuli sciolti di istiociti vicino ai bronchioli. La granulomatosi con poliangioite (in passato chiamata anche granulomatosi di Wegener) è caratterizzata da necrosi basofila circondata da un infiltrato cellulare contenente cellule giganti. La granulomatosi sarcoide nodulare è talvolta considerata una variante della sarcoidosi ed è anch’essa caratterizzata da una necrosi estesa che, a differenza della granulomatosi con poliangioite, è eosinofila e delimitata da numerosi granulomi compatti accompagnati da vasculite granulomatosa senza distruzione delle pareti dei vasi.
La diagnosi differenziale in caso di manifestazioni neurologiche deve basarsi sui risultati della risonanza magnetica (soprattutto lesioni focali periventricolari rispetto a lesioni parenchimali o meningee). Le nuove tecniche di risonanza magnetica hanno ottimizzato la sensibilità, ma a causa della mancanza di specificità, il contenimento diagnostico rimane una sfida. [2]. Lo spettro delle diagnosi differenziali spazia dalle malattie autoimmunologiche, infiammatorie o idiopatiche (ad esempio, SM, malattia dello spettro della neuromielite optica, LES, sindrome di Sjögren, malattia di Behcet, vasculite primaria del SNC) alle entità infettive (ad esempio, tubercolosi, malattia di Lyme, neurosifilide, tassoplasmosi) e alle neoplasie (neoplasie primarie del SNC, linfomi e altre) [2].
Letteratura:
- «Sarcoidosis: Beyond the Lungs and Lymph Nodes», Prof. Dr. med. Jörg D. Seebach, Allergy and Immunology Update, Grindelwald, 27.–29.1.2023.
- Franzen DP, et al.: Sarcoidosis – a multisystem disease. Swiss Med Wkly. 2022;152: w 30049.
- Graf L, Geiser T: Das Chamäleon unter den Systemerkrankungen: «Die Sarkoidose». Swiss Med Forum 2018; 18(35): 695–701.
- Valeyre D, et al.: Sarcoidosis. Lancet (London, England) 2014; 383(9923): 1155–1167.
- Lichtenberger N: Diagnostik und Therapie der Kardialen und pulmonalen Sarkoidose. https://opus.bibliothek.uni-wuerzburg.de, (letzter Abruf 23.02.2023)
- Costabel U, et al. [Cardiac sarcoidosis: diagnostic and therapeutic algorithms]. Pneumologie (Stuttgart, Germany) 2014; 68(2): 124–132.
- Birnie DH, et al.: HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart rhythm 2014; 11(7): 1305–1323.
- Giblin GT, et al.: Cardiac Sarcoidosis: When and How to Treat Inflammation. Card Fail Rev 2021;7:e17. doi: 10.15420/cfr.2021.16.
- Hamzeh N, et al.: Pathophysiology and clinical management of cardiac sarcoidosis. Nat Rev Cardiol 2015; 12(5): 278–288.
- Valeyre D, et al.: Clinical presentation of sarcoidosis and diagnostic work-up. Semin Respir Crit Care Med 2014; 35(3): 336–351.
- Spagnolo P, et al.: Pulmonary sarcoidosis. Lancet Respir Med 2018; 6(5): 389–402.
- Grunewald J, et al.: Sarcoidosis. Nat Rev Dis Primers 2019; 5(1): 45.
- Birnie D, et al.: Cardiac Sarcoidosis. Clinics in chest medicine 2015; 36(4): 657–668.
HAUSARZT PRAXIS 2023; 18(5): 40–42
Foto di copertina: Hellerhoff, wikimedia