La malattia polmonare interstiziale è relativamente rara nella medicina generale. Tuttavia, dovrebbe essere presa in considerazione nei casi di dispnea cronica, tosse secca e rantoli fini e spumeggianti. La diagnosi precoce e corretta è fondamentale.
Le malattie polmonari interstiziali (ILD) comprendono un ampio gruppo di diverse patologie rare del parenchima polmonare con una presentazione clinica, radiologica e patologica simile [1]. I processi alla base della ILD, la prognosi, ma anche le terapie differiscono notevolmente da entità a entità. Da un lato, troviamo malattie con una prognosi favorevole, ma anche malattie con un decorso acuto e un’elevata mortalità. La fibrosi polmonare idiopatica (IPF), la più comune ILD tra le polmoniti interstiziali idiopatiche (IIP), ha un decorso paragonabile a quello dei tumori maligni aggressivi, con una sopravvivenza mediana di circa tre anni dopo la diagnosi [2].
Il trattamento dell’ILD nei singoli pazienti dipende in gran parte dall’entità della malattia. Per alcuni modelli di malattia, l’attenzione si concentra sull’astinenza dall’esposizione a sostanze nocive (inalatorie). A seconda dell’entità, si utilizzano terapie immunosoppressive o, nel caso dell’IPF, terapie antifibrotiche. Una diagnosi precoce e corretta, con particolare riconoscimento dei fattori reversibili, è fondamentale per una consulenza e un trattamento ottimali dei pazienti.
Classificazione delle malattie polmonari interstiziali
L’ILD può essere suddivisa in malattie con una causa nota e malattie con una causa sconosciuta (polmonite interstiziale idiopatica, IIP) (Fig. 1) . L’elenco delle possibili cause di ILD è lungo. Tra le cause note, troviamo l’esposizione a polveri inorganiche o organiche, a farmaci e a raggi X. L’ILD complicata può verificarsi in molte malattie reumatologiche. L’ILD può essere la prima manifestazione di una malattia reumatologica sottostante, prima che possa essere diagnosticata con i consueti criteri diagnostici. Nel 2015 è stato introdotto il termine “polmonite interstiziale con caratteristiche autoimmuni” (IPAF) per riflettere questa situazione [3].
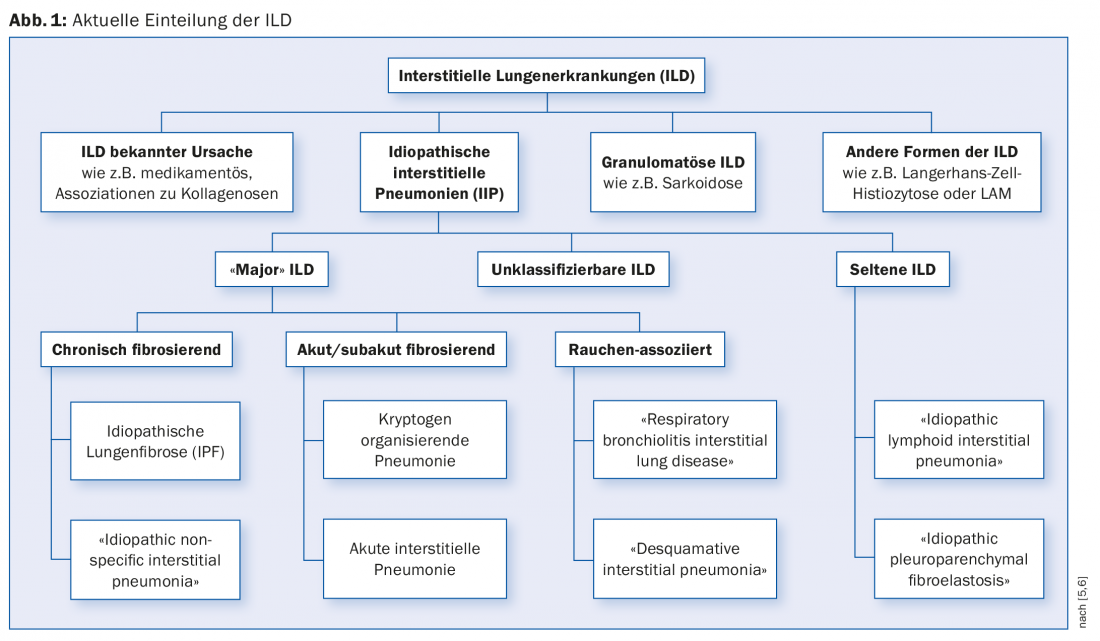
La causa dell’IIP è, come suggerisce il nome, poco chiara. Le IIP “principali” sono classificate in base al decorso clinico in fibrosi cronica, fibrosi acuta/subacuta e malattie associate al fumo.
La denominazione “idiopatica” è in parte contraddittoria rispetto alla fisiopatologia del gruppo di malattie associate al fumo, pertanto si discute una categorizzazione al di fuori dell’IIP. Il fumo provoca cambiamenti istologicamente rilevabili (“bronchiolite respiratoria”). I soggetti sensibili possono sviluppare ILD (“bronchiolite respiratoria interstiziale” [RB-ILD] e “polmonite interstiziale desquamativa”) [4].
Epidemiologia della ILD
Le ILD sono malattie rare, di conseguenza ci sono pochi dati epidemiologici. Un ampio studio epidemiologico del 1994 ha rilevato un’incidenza di 31,5 casi per 100.000/anno negli uomini e di 26,1 casi per 100.000/anno nelle donne [7]. L’IPF, più comune negli uomini anziani, è l’entità più comune tra le IILD. Non esistono dati epidemiologici sulla IPF in Svizzera. Si stima che ci siano da 100 a 5000 pazienti con IPF in Svizzera (prevalenza 1,25-63 casi/100.000) [8]. L’età della prima diagnosi di ILD varia tra le diverse entità patologiche. Mentre l’incidenza dell’IPF aumenta significativamente con l’età, le sarcoidosi, le istiocitosi a cellule di Langerhans o, nelle donne, le linfangioleiomiomatosi (LAM) sono più comuni nella giovane età adulta.
Diagnosi di ILD
La diagnosi di ILD viene effettuata tenendo conto dei risultati clinici, radiologici, della funzionalità polmonare, della chimica di laboratorio e, a seconda della situazione, dei risultati citologici/istologici. La diagnosi e la determinazione della strategia terapeutica per il singolo paziente vengono effettuate in modo ottimale in un contesto interdisciplinare composto da medici (pneumologi), radiologi e patologi (commissione ILD). È stato dimostrato che è possibile ottenere un significativo miglioramento qualitativo della diagnostica [9].
Clinica: la clinica dell’ILD è spesso aspecifica, con dispnea in lento aumento e di solito tosse secca. L’esame clinico spesso rivela rantoli a bolle fini accentuati al basale e, in particolare nella malattia avanzata, una saturazione dell’ossigeno ridotta (soprattutto durante lo sforzo) e segni di ipossiemia cronica, come le unghie a vetro di orologio e le dita a bacchetta di tamburo. È importante considerare la presenza di ILD nelle prime fasi della diagnosi differenziale dei pazienti con questi sintomi.
Spesso, un’anamnesi accurata ci fornisce indizi decisivi sull’entità dell’ILD. Sono importanti la durata dei sintomi, ma anche le possibili influenze ambientali, compresa un’anamnesi professionale dettagliata. L’identificazione delle influenze ambientali potenzialmente reversibili è di grande importanza. Il riconoscimento di possibili aeroallergeni che possono scatenare una polmonite da ipersensibilità (alveolite allergica esogena, EAA) è elementare sia per la diagnosi che per la bonifica decisiva dell’esposizione. Il più delle volte si tratta di allevamento di uccelli, contatto con l’agricoltura o esposizione a muffe. Il fumo è un fattore scatenante potenzialmente reversibile nell’ILD associata al fumo. Oltre alla malattia pleurica associata all’amianto, l’esposizione all’amianto può anche portare alla fibrosi polmonare associata all’amianto. I sintomi di una malattia reumatologica sottostante devono essere ricercati in modo specifico. Molti farmaci possono causare l’ILD come effetto collaterale. A questo punto, vorremmo fare riferimento alla homepage www.pneumotox.com, che è molto utile nella vita quotidiana e fornisce informazioni sugli effetti collaterali tossici per i polmoni noti.
È necessario raccogliere l’anamnesi familiare; fino al 20% di tutti i casi di ILD presenta una forma familiare con mutazioni genetiche corrispondenti [10].
Imaging: la radiografia convenzionale del torace spesso suggerisce una malattia polmonare interstiziale con un aumento dei segni reticolari e/o nodulari del polmone.
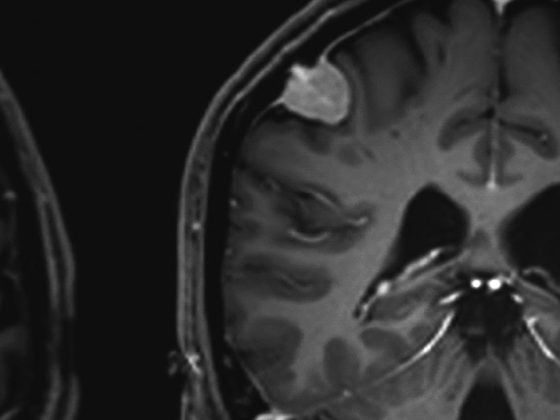
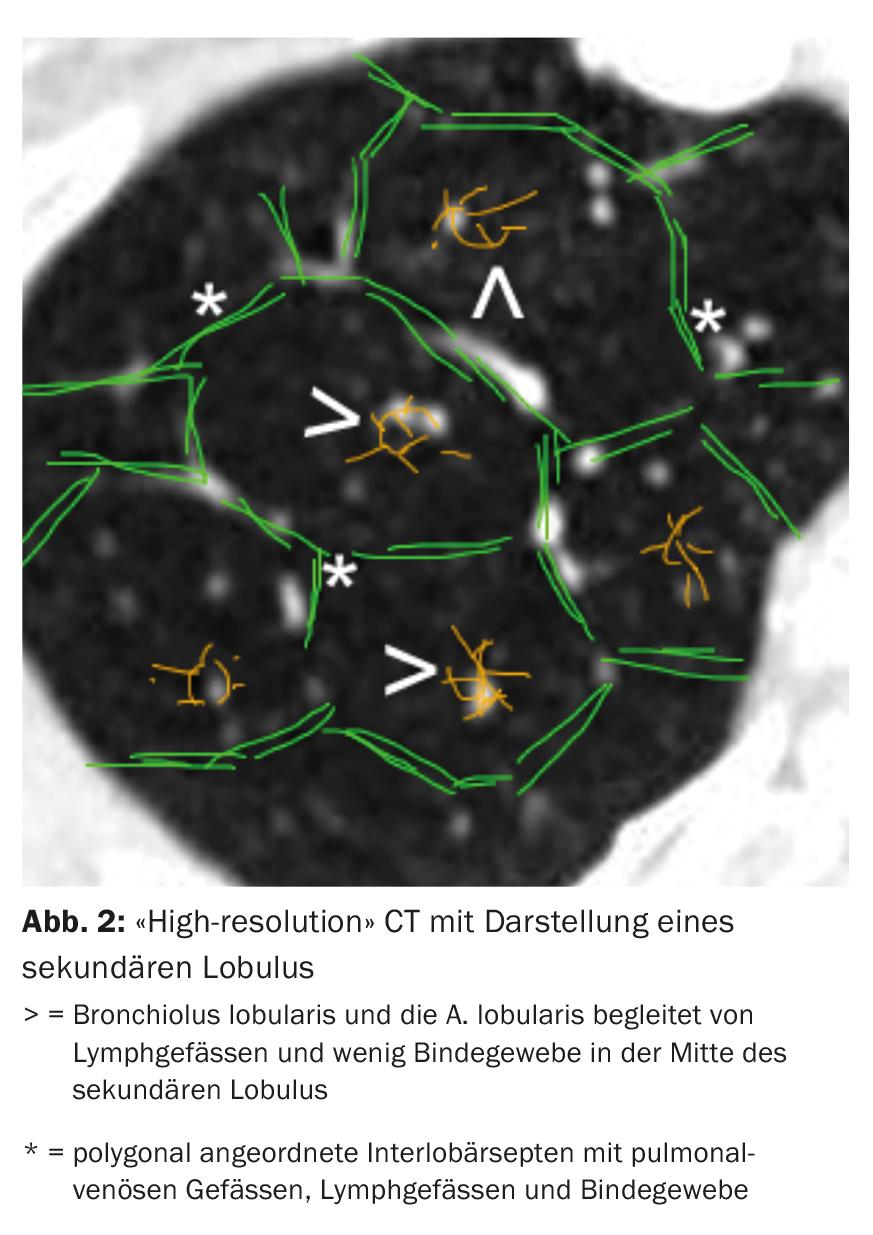
La tomografia computerizzata (TC) del torace con fette ad “alta risoluzione” svolge un ruolo centrale nella diagnosi di ILD. In base al tipo e alla disposizione delle alterazioni in relazione alla localizzazione del lobulo secondario (Fig. 2) e alla topografia polmonare, si possono distinguere diversi modelli radiologici.
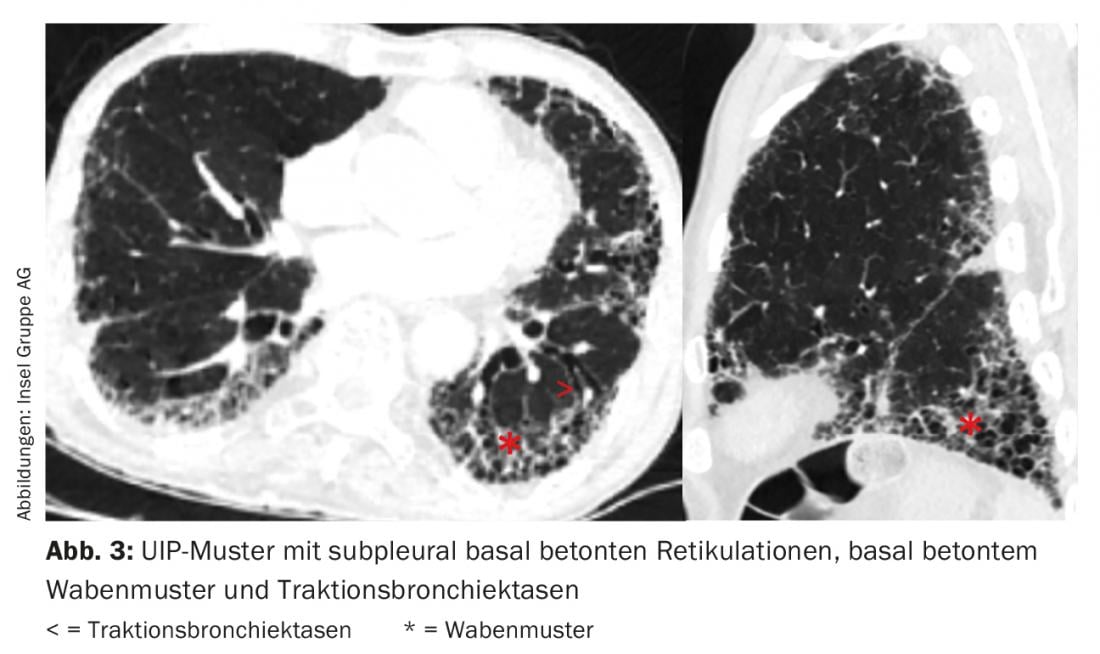
Il singolo pattern radiologico non è specifico per un’entità di ILD. Per esempio, nell’IPF troviamo un modello di “polmonite interstiziale abituale” (UIP) con reticoli subpleurici accentuati al basale, modello a nido d’ape accentuato al basale e bronchiectasie da trazione (Fig. 3) [11]. Tuttavia, un modello UIP può essere riscontrato anche in altre entità, come la vasculite dei piccoli vasi, l’EAA cronica, l’ILD nel contesto dell’artrite reumatoide o dell’asbestosi.
Funzione polmonare: nell’ILD di solito troviamo una restrizione e un’alterazione dello scambio di gas (capacità di diffusione). Con il progredire della malattia, si verifica inizialmente un’ipossiemia indotta dall’esercizio fisico e, con il progredire della malattia, un’ipossiemia indotta dal riposo.
La capacità vitale forzata (FVC) viene utilizzata come importante parametro di progressione. Un calo di FVC ≥10% in 24 settimane aumenta il rischio di mortalità nei dodici mesi successivi di un fattore di 4,8 [12]. Oltre alla FVC facilmente misurabile, anche la capacità di diffusione, il test del cammino di 6 minuti e gli indici multivariabili sono correlati alla prognosi [13].
Laboratorio: il laboratorio può essere utile per classificare l’ILD. Oltre all’emocromo (eosinofilia), la funzionalità renale, i valori epatici e infiammatori, nonché la ricerca sierologica di una malattia reumatologica sottostante possono darci indicazioni.
Indagini invasive: Di norma, nei pazienti con una nuova diagnosi di ILD viene eseguita una broncoscopia con almeno un lavaggio broncoalveolare (BAL). Un BAL sempre più emorragico da una porzione all’altra è tipico dell’emorragia alveolare (ad esempio, nella vasculite dei piccoli vasi). La distribuzione cellulare nel BAL mostra cambiamenti caratteristici in alcune entità ILD (ad esempio, eosinofilia nella polmonite eosinofila, linfocitosi nella sarcoidosi e nella EAA). A seconda del sospetto diagnostico basato sulla presentazione clinica e radiologica, vengono eseguite biopsie linfonodali mediastiniche/ilari e/o biopsie polmonari transbronchiali. I frammenti di tessuto delle biopsie polmonari transbronchiali sono spesso troppo piccoli per una diagnosi affidabile. Campioni di tessuto più grandi possono essere ottenuti con la toracoscopia o, da qualche anno, anche per via broncoscopica con la criobiopsia.
Terapia
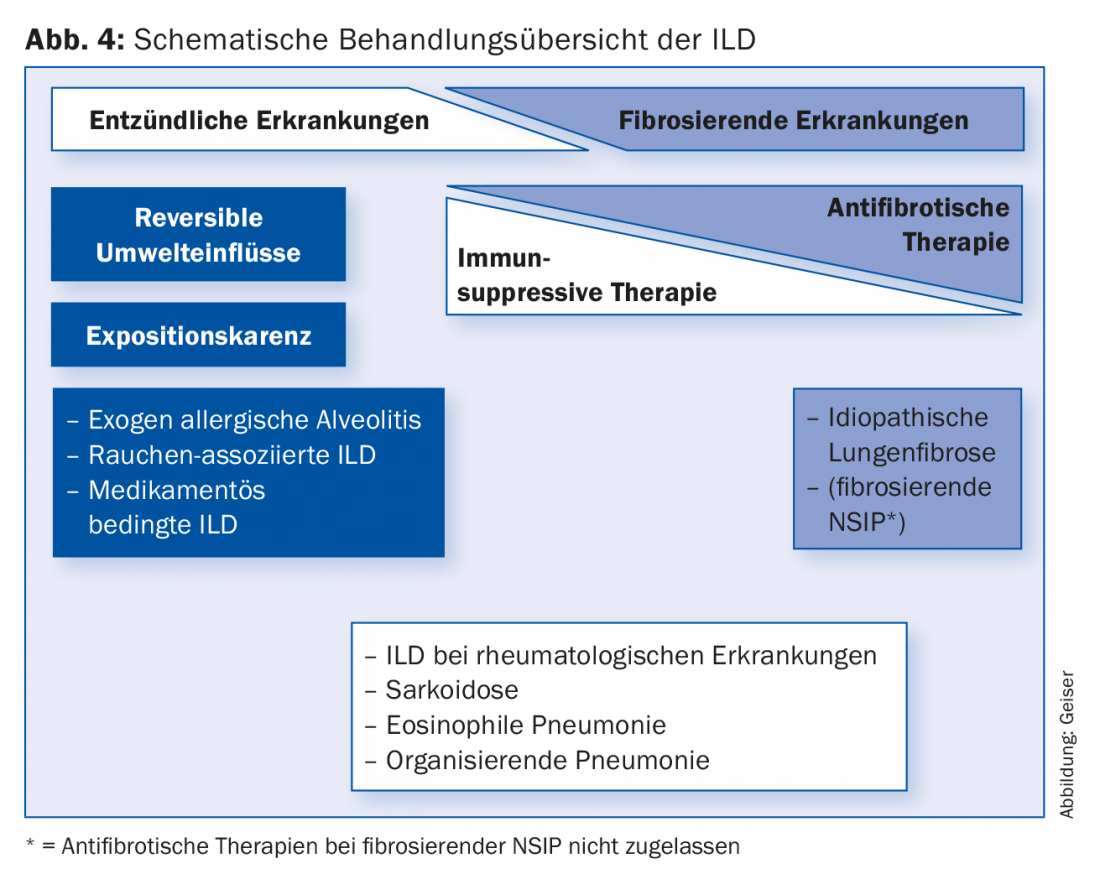
La terapia dell’ILD differisce fondamentalmente da entità a entità (Fig. 4). In alcune entità, ad esempio, la malattia può essere influenzata in modo decisivo dall’astinenza dall’esposizione (cessazione del fumo, astinenza dai fattori scatenanti dell’alveolite allergica esogena, cambio di farmaci). Molte ILD sono trattate con l’immunosoppressione, come le malattie associate alle collagenosi/vasculiti, le polmoniti organizzative (criptogenetiche), le polmoniti eosinofile o le sarcoidosi. L’immunosoppressione viene solitamente effettuata con corticosteroidi sistemici, spesso in combinazione con un farmaco che risparmia gli steroidi (ad esempio, azatioprina o micofenolato).
Le terapie immunosoppressive comunemente utilizzate per l’IPF fino a pochi anni fa non sono più raccomandate [14,15]. Dopo molti studi negativi con varie sostanze, i due farmaci attualmente approvati per il trattamento dell’IPF, pirfenidone (Esbriet®) e nintedanib (Ofev®), hanno dimostrato di rallentare la progressione della malattia [16–18]. Nuovi dati suggeriscono che la sopravvivenza può essere influenzata favorevolmente.
Nei pazienti più giovani con entità ILD a rapida progressione, come l’IPF, la valutazione per il trapianto di polmone dovrebbe avvenire precocemente nel decorso della malattia. Con il progredire dell’ILD, può essere necessaria un’ossigenoterapia a lungo termine per i pazienti ipossiemici e ulteriori terapie palliative, compresa la riabilitazione polmonare.
Messaggi da portare a casa
- Sebbene la malattia polmonare interstiziale (ILD) sia relativamente poco frequente nella medicina generale, nei casi di dispnea cronica, secchezza
- La tosse e i rantoli fini devono essere considerati come diagnosi differenziale.
- Tra le ILD, troviamo entità patologiche molto diverse, con decorsi diversi.
- Una diagnosi precoce e corretta è fondamentale per una consulenza e un trattamento ottimali dei pazienti.
- La terapia dell’ILD varia in modo significativo a seconda dell’entità della malattia e va dall’astinenza dall’esposizione agli agenti scatenanti all’immunosoppressione e all’uso di terapie antifibrotiche nella fibrosi polmonare idiopatica (IPF).
- I pazienti più giovani con un’entità di ILD in rapida progressione dovrebbero essere valutati per il trapianto di polmone in una fase precoce.
- Nel corso del decorso, potrebbe rendersi necessaria un’ossigenoterapia a lungo termine nei pazienti ipossiemici e ulteriori terapie palliative.
Letteratura:
- Cosgrove G, Schwarz M: Approccio alla valutazione e alla diagnosi della malattia polmonare interstiziale. In: Schwarz M, King T (eds.): Malattia polmonare interstiziale. 5a ed. Shelton: People’s Medical Publishing House-USA 2011: 3-34.
- Latsi PI, et al: Polmonite interstiziale idiopatica fibrotica: il valore prognostico dei trend funzionali longitudinali. Am J Respir Crit Care Med 2003; 168(5): 531-537.
- Fischer A, et al: Una dichiarazione di ricerca ufficiale della European Respiratory Society/American Thoracic Society: polmonite interstiziale con caratteristiche autoimmuni. Eur Respir J 2015; 46(4): 976-987.
- Fraig M, et al: Bronchiolite respiratoria: uno studio clinicopatologico in fumatori attuali, ex fumatori e mai fumatori. Am J Surg Pathol 2002; 26(5): 647-653.
- American Thoracic Society/European Respiratory Society: Classificazione internazionale di consenso multidisciplinare delle polmoniti interstiziali idiopatiche. Am J Respir Crit Care Med 2002; 165(2): 277-304.
- Travis WD, et al: Una dichiarazione ufficiale dell’American Thoracic Society/European Respiratory Society: Aggiornamento della classificazione multidisciplinare internazionale delle polmoniti interstiziali idiopatiche. Am J Respir Crit Care Med 2013; 188(6): 733-748.
- Coultas DB, et al: L’epidemiologia delle malattie polmonari interstiziali. Am J Respir Crit Care Med 1994; 150(4): 967-972.
- Funke M, Geiser T: Fibrosi polmonare idiopatica: la svolta è ora! Swiss Med Wkly 2015; 145: w14139.
- Flaherty KR, et al: Polmonite interstiziale idiopatica: qual è l’effetto di un approccio multidisciplinare alla diagnosi? Am J Respir Crit Care Med 2004; 170(8): 904-910.
- Kropski JA, Blackwell TS, Loyd JE: La base genetica della fibrosi polmonare idiopatica. Eur Respir J 2015; 45(6): 1717-1727.
- Raghu G, et al: Una dichiarazione ufficiale ATS/ERS/JRS/ALAT: fibrosi polmonare idiopatica: linee guida basate sull’evidenza per la diagnosi e la gestione. Am J Respir Crit Care Med 2011; 183(6): 788-824.
- du Bois RM, et al: Capacità vitale forzata nei pazienti con fibrosi polmonare idiopatica: proprietà del test e differenza minima clinicamente importante. Am J Respir Crit Care Med 2011; 184(12): 1382-1389.
- Sharp C, Adamali HI, Millar AB: Un confronto degli indici multidimensionali pubblicati per prevedere l’esito nella fibrosi polmonare idiopatica. ERJ Open Res 2017; 3(1). DOI: 10.1183/23120541.00096-2016.
- Funke M, et al: Fibrosi polmonare idiopatica in Svizzera: diagnosi e trattamento. Respirazione 2017; 93(5): 363-378.
- Raghu G, et al: Una linea guida ufficiale di pratica clinica ATS/ERS/JRS/ALAT: Trattamento della fibrosi polmonare idiopatica. Un aggiornamento della linea guida di pratica clinica del 2011. Am J Respir Crit Care Med 2015; 192(2): e3-19.
- Noble PW, et al: Pirfenidone nei pazienti con fibrosi polmonare idiopatica (CAPACITY): due studi randomizzati. Lancet 2011; 377(9779): 1760-1769.
- Richeldi L, et al: Efficacia e sicurezza di nintedanib nella fibrosi polmonare idiopatica. N Engl J Med 2014; 370(22): 2071-2082.
- King TE Jr, et al: Uno studio di fase 3 sul pirfenidone nei pazienti con fibrosi polmonare idiopatica. N Engl J Med 2014; 370(22): 2083-2092.
PRATICA GP 2017; 12(12): 13-18